All published articles of this journal are available on ScienceDirect.

Changes in Coagulation and Platelet Reactivity in People with HIV-1 Switching Between Abacavir and Tenofovir
Authors Info & Affiliations
Abstract
Background:
Several studies have shown an association between abacavir (ABC) and increased risk of myocardial infarction (MI), but the causative mechanism has not been established. Both vascular endothelial inflammation and platelet activation have been proposed as contributing factors.
Objective:
The study aims to investigate the effects of ABC relative to tenofovir disoproxil (TDF) on functional assays of primary and secondary hemostasis and a comprehensible range of relevant biomarkers.
Methods:
In an investigator-initiated, open-labeled, crossover trial, we included HIV-infected males receiving either ABC or TDF and switched treatment to the alternate drug. At inclusion and after three months on the new regimen, we performed Multiplate® and thromboelastography (TEG®) and measured biomarkers of coagulation, inflammation, platelet reactivity, endothelial disruption and activation, and fibrinolysis, lipids, HIV RNA, CD4, CD8, and creatinine. Treatment effects were assessed by comparing intraindividual differences between the two treatment orders by the Wilcoxon Rank Sum test.
Results:
In total, 43 individuals completed the study. No intraindividual differences were observed for Multiplate® or TEG® when switching between regimens. We observed a significant treatment effect on coagulation factors II-VII-X (p<0.0001), sCD40L (a biomarker of platelet reactivity, p=0.04), thrombomodulin (biomarker of endothelial damage, p=0.04), lipids, and CD8 cell counts (p=0.04), with higher values during ABC treatment compared to TDF.
Conclusion:
Compared to TDF, ABC treatment affected several outcome measures in a pro-coagulant direction. Suggesting that the risk of MI associated with ABC may be caused by the sum of multiple, discrete disturbances in the hemostatic system and endothelium.
Study Registration:
The trial was registered at clinicaltrials.gov (NCT02093585).
1. INTRODUCTION
The nucleoside reverse transcriptase inhibitor (NRTI) abacavir (ABC) was approved by the European Medicine Agency in 1999 (Ziagen®). Nine years later, a possible association between ABC and myocardial infarction (MI) was presented by The Data Collection on Adverse events of Anti-HIV Drugs (D:A:D) study group [1]. The large, prospective observational cohort included 33,347 people with HIV (PWH). Recent use of ABC was associated with an increased rate of MI (RR 1.90, 95% CI 1.47-2.45). The association remained after adjustment for cardiovascular risk factors, predicted 10-year risk of coronary heart disease, and adjustment to overcome prescription bias [2, 3]. The excess risk subsided 6 months after drug cessation, indicating a reversible mechanism [1]. In contrast, the NRTI tenofovir disoproxil (TDF) does not appear to have the same effect.
Since then, numerous studies have addressed the topic with conflicting results [4-8]. A systematic review and meta-analysis from 2018 found that PWH with recent exposure to ABC had approximately 60% increased risk of cardiovascular disease (CVD) compared to PWH receiving ABC-free regimens [9].
The biological mechanism explaining the observed association between ABC and MI has not been fully established. Different pathophysiological explanations have been proposed and investigated. Vascular endothelial inflammation and activation of platelets have been suggested as primary contributors to thrombosis, as reviewed [10, 11].
The objective of this study was to investigate the effects of ABC relative to TDF on primary and secondary hemostasis and biomarkers of coagulation, inflammation, platelet reactivity, endothelial disruption and activation, and fibrinolysis using a cross-over study design.
2. MATERIALS AND METHODS
2.1. Study Design
We conducted an investigator-initiated, open-labeled, crossover trial including HIV-1-infected individuals on combination antiretroviral therapy (cART) containing either TDF or ABC. The study design is illustrated in (Fig. 1). Participants had blood drawn at inclusion and then switched to the alternative treatment regimen. The group of individuals switching from ABC to TDF containing regimens was termed “AT,” and the group of individuals switching from TDF to ABC was termed “TA”. For some, switching ABC for TDF and vice versa also meant switching lamivudine for emtricitabine and vice versa. Lamivudine and emtricitabine are considered analogous and can be interchanged without restriction [12]. Individuals who were treated with a Kivexa® (ABC+lamivudine) based regimen were switched to Truvada® (TDF+ emtricitabine). Individuals treated with either a Truvada® (TDF+emtricitabine), Atripla® (efavirenz+ emtricitabine+TDF) or Viread® (TDF) + Epivir® (lamivudine) based regimen were switched to Kivexa® (ABC+ lamivudine). The third drug in the combination remained unchanged. The new regimen was continued for 85-90 days before participants had blood drawn again. Hereafter, the participants could continue the new regimen or switch back to their original regimen as they preferred.
2.2. Ethics Approval and Consent to Participate
Participants had to be able to understand and sign oral and written informed consent and were recruited in connection with their routine visits. The trial was approved by the Danish Data Protection Agency (30-0981), the Ethical Committee for Biochemical Research in the Capital Region, Denmark (H-2-2013-082), and by the Danish Health and Medicines Authority (2013051722). The trial was registered at clinicaltrials.gov (NCT02093585) and at clinicaltrialsregister.eu (EudraCT 2013-001685-42).
2.3. Participants
Eligible participants were identified from the Danish HIV-cohort, including all PWH treated in Denmark after 31 December 1994 [13]. We identified individuals who were treated at the Department of Infectious Diseases at Rigshospitalet, University Hospital of Copenhagen, male, 35 to 70 years old, hepatitis B and C negative, and treated with either TDF or ABC >6 months at the time of inclusion. Medical charts of these individuals were manually assessed for eligibility using the following exclusion criteria; HIV-2-infection, HIV RNA ≥400 copies/mL within 6 months prior to inclusion, previous ischemic heart disease, peripheral atherosclerotic disease or stroke, currently receiving anticoagulant therapy, aspirin or NSAIDs, known coagulation disorder, platelet count <150x109/L within 6 months prior to inclusion, eGFR < 70 mL/min within 6 months prior to inclusion, HLA-B*5701 genotype [14] or known allergy towards ABC or TDF. The trial inclusion occurred from February 2014 until January 2015.
We aimed to include 100 PWH; however, fewer individuals than expected wished to participate. Therefore, we decided to end recruitment after 43 participants completed the study.
2.4. Outcomes
The outcomes were changes in primary and secondary hemostasis proxied by platelet aggregation by Multiplate® and clot formation by thromboelastographic (TEG®), respectively. Furthermore, we assessed changes in lipids and biomarkers of coagulation, inflammation, platelet reactivity, endothelial disruption and activation, and fibrinolysis, as specified below. HIV-RNA, CD4 and CD8 cell counts, creatinine, and eGFR were included as treatment efficacy and toxicity measures.
At baseline, age, sex, race, medications other than antiretroviral drugs, compliance, and current smoking status were registered.
2.5. Laboratory Methods
Platelet aggregation was evaluated in heparinized whole blood by impedance aggregometry using Multiplate® Analyzer (Dynabyte GmbH/Roche Diagnostics) and applying commercially available platelet agonists according to the manufacturer’s recommendations. Tests performed were; TRAP test (thrombin receptor-activating peptide TRAP-6 32 µM), ADP test (adenosine diphosphate ADP 6.5 µM), ASPI test (arachidonic acid 0.5 µM), RISTO low test (ristocetin 0.2 mg/mL), and RISTO high test (ristocetin 0.77 mg/mL). Results were displayed as the area under the curve (AUC) and expressed as aggregation units (U). Reference values for U were reported by the manufacturer.
Clot formation was assessed in kaolin induced re-calcified citrated whole blood by TEG® 5000 Thromboelastograph® Hemostasis Analyzer System (Haemonetics Corp. Braintree, MA) according to the manufacturer’s recommendations. All analyses were conducted at 37oC. The variables recorded were reaction time (R), rate of initial fibrin formation (Angle), maximum amplitude (MA), and lysis after 30 minutes (Ly30).
Biomarkers were analyzed by commercially available enzyme-linked immunosorbent assay (ELISA), according to the manufacturer’s recommendations. The following assays were applied: Syndecan-1 (Nordic Biosite/Diaclone), Thrombomodulin (Nordic Biosite/Diaclone), soluble E-selectin (sE-selectin) (Nordic Biosite/Diaclone), soluble CD40 ligand (sCD40L) (R&D Systems), interleukin-6 (IL-6) (R&D Systems D6050), Transforming growth factor beta (TGF-β) (R&D Systems), Beta thromboglobulin (USCN/biotech SEA370Hu), Platelet factor 4 (USCN/biotech SEA172Hu), Soluble glycoprotein VI (sGPVI) (USCN/biotech SEB904Hu), plasminogen activator inhibitor (PAI)-1 (Sekisui diag/BioNordika), and tissue-type plasminogen activator (tPA) (Technozym/Bionordika).
HIV-RNA was measured by COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Test (Roche). CD4 and CD8 cell counts were measured by the single platform lyse-no-wash procedure, using BD (Becton, Dickinson and company) TRUcount beads and monoclonal antibodies against CD3, CD4, and CD8. Total cholesterol, high density lipids (HDL), low density lipids (LDL), triglycerides, creatinine, activated partial thromboplastin time (APTT), coagulation factor II-VII-X, platelet count, fibrinogen, D-dimer, antithrombin (ATIII), and high sensitivity C-reactive protein (hs-CRP) were measured by standard methods in the hospital’s ISO 15189 certified Department of Biochemistry.
Samples for TEG and platelet aggregation analyses were analyzed immediately. Additional markers of coagulation, HIV status, and lipids were sent to routine analysis directly after the sample was drawn. Samples for analyses of markers of inflammation, platelet reactivity, endothelial damage, and fibrinolysis were sent to the laboratory, where the plasma was frozen and stored, and analyses were performed in bulk.
2.6. Statistics
All results are presented as medians and interquartile ranges (IQR) or absolute number and percentage (n (%)). Treatment effects were assessed using the Wilcoxon Rank Sum test, comparing intra-individual (within-person) differences between the two time points in the results of Multiplate®, TEG®, and biomarkers between treatment groups (AT and TA) [15].
Values below or higher than the respective kit lower/higher detection limits were set to the value of the lower/higher detection limit to generate the most conservative estimate. Samples that could not be analyzed (e.g., wrong test tubes or insufficiently filled test tubes) were not repeated and the person was excluded from the specific analysis. Numbers included in each analysis are specified in Table 1. P-values < 0.05 were considered statistically significant. Calculations were performed using StataCorp. 2017. Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC and SAS Enterprise Guide software, Version 7.15 HF7. Copyright © 2017, SAS Institute Inc., Cary, NC, USA.
3. RESULTS
3.1. Participants
In total, 55 PWH consented to participate, of whom 21 received ABC at inclusion and entered the AT group switching from ABC to TDF, while 34 received TDF at inclusion and entered the TA group switching from TDF to ABC. In the AT group, one person was (per protocol) excluded because of HLA-B*5701 genotype and one person dropped out due to unacceptable side effects. In the TA group, one person was excluded because of the HLA-B*5701 genotype, eight dropped out due to unacceptable side effects, and one was lost to follow-up. Thus, 43 individuals completed the study, 19 in the AT group and 24 in the TA group. Participant flow is presented in Fig. (1), and baseline characteristics are shown in Table 1.
3.2. Outcomes
Results of Multiplate® and TEG® are presented in Table 2 and Supplementary Figs. (1 and 2). No significant treatment effects were found on primary and secondary hemostasis assessed with Multiplate® or TEG®.

Both total cholesterol, HDL, and LDL were significantly higher when participants were receiving ABC compared to TDF (Supplementary Table 1 and Supplementary Fig. 3). The median intraindividual change in the AT group vs. TA group was -23 mg/dL (IQR:-35-8) vs. 31 mg/dL (IQR: 8-48), p <0.0001 for cholesterol, -5 mg/dL (IQR:-8--1) vs. 4 mg/dL(IQR:-1-12) (, p=0.0017 for HDL, and -15 mg/dL (IQR:-27-0) vs. 25 mg/dL (IQR:6-35) for LDL. Levels of triglycerides did not differ between treatment regimens; median 5 mg/dL (IQR: -54-29) vs 11 mg/dL (IQR: -23-31), p 0.58. No changes were observed in the inflammation biomarkers, IL-6 and hs-CRP (Supplementary Table 2 and Supplementary Fig. 4).
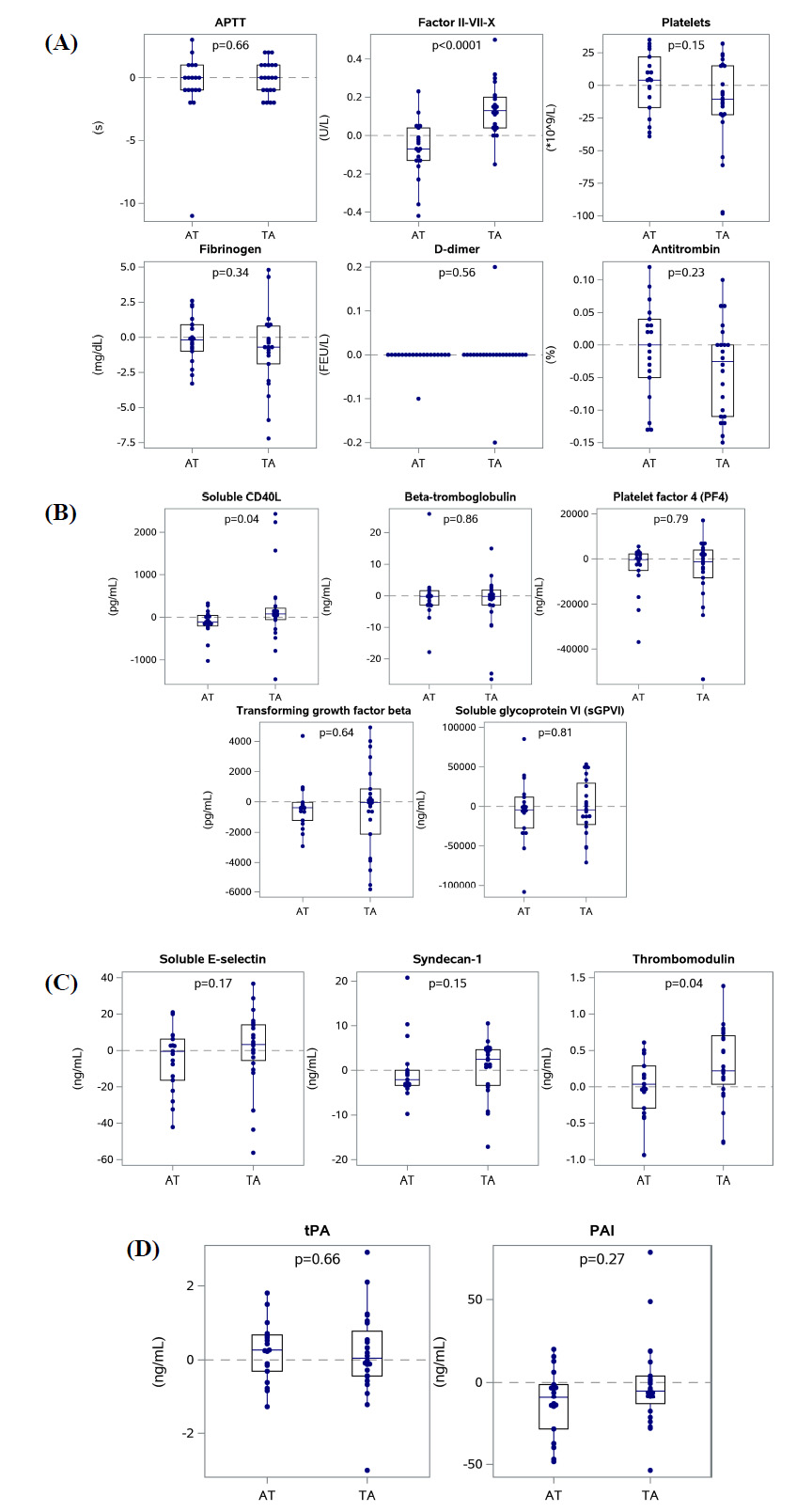
Results of biomarkers of coagulation, platelet reactivity, endothelial disruption and activation, and fibrinolysis are in Table 3 and Fig. (2A-D). Normal ranges are included where established. Coagulation factor II-VII-X levels, sCD40L (a biomarker of platelet reactivity), and thrombomodulin (a biomarker of endothelial damage) were higher during treatment with ABC than TDF. No differences in intraindividual changes were observed for the remaining biomarkers (Table 3).
| - |
AT Group n=19 |
TA Group n=24 |
Total n=43 |
p-value* |
|---|---|---|---|---|
| Age at inclusion, years (median (range)) | 50.1 (39-65) | 48.8 (37-69) | 49.4 (37-69) | 0.32 |
| Years since HIV-diagnosis (median (range)) | 16 (3-30) | 9 (2-30) | 11 (2-30) | 0.12 |
| Years on HIV-treatment (median (range)) | 13 (3-21) | 8 (2-19) | 10 (2-21) | 0.16 |
| Male sex, n (%) | 19 (100) | 24 (100) | 43 (100) | . |
| Caucasian, n (%) | 17 (89.5) | 23 (95.8) | 42 (97.7) | 0.57 |
| Active smoker at inclusion, n (%) | 6 (31.6) | 12 (50) | 18 (41.9) | 0.22 |
| Poor compliance (patients with > 3 doses not taken the previous month) n (%) | 0 (0) | 0 (0) | 0 (0) | . |
| - | Normal Range | AT Group | TA Group | p-Values | ||||
|---|---|---|---|---|---|---|---|---|
| N | Baseline (ABC Treatment) | Intra-Individual Change when Switching from ABC to TDF | N | Baseline (TDF Treatment) | Intra-Individual Change when Switching from TDF to ABC | Wilcoxon Rank Sum | ||
| 90%CI | Median (IQR) | Median (IQR) | Median (IQR) | Median (IQR) | ||||
| Primary and Secondary Hemostasis | ||||||||
| Multiplate® | ||||||||
| TRAP (U) | 92-151 | 19 | 108 (92-122) | -3 (-23-13) | 24 | 109 (92-118) | -2 (-13-14) | 0.32 |
| ADP (U) | 55-117 | 19 | 78 (65-90) | 1 (-16-11) | 24 | 79 (66-90) | -3 (-14-13) | 0.93 |
| ASPI (U) | 79-141 | 19 | 84 (71-103) | -2 (-7-6) | 24 | 88 (71-109) | -1 (-9-5) | 0.87 |
| RISTO LOW (U) | 13-69 | 18 | 31 (23-41) | -2 (-15-5) | 23 | 25 (21-36) | 1 (-3-5) | 0.42 |
| RISTO HIGH (U) | 65-116 | 18 | 76 (63-88) | -2 (-13-9) | 23 | 86 (71-94) | 5 (-19-13) | 0.79 |
| TEG® | ||||||||
| R (min) | 3-9 | 19 | 6.9 (6.2-8.5) | -0.7 (-1.1-0.6) | 24 | 6.8 (6.0-7.9) | -0.1 (-1.1-0.9) | 0.50 |
| Angle (degrees) | 55-75 | 19 | 63.8 (61.0-67.5) | 0.0 (-6.5-4.7) | 24 | 64.6 (60.4-67.9) | 1.3 (-3.2-10.4) | 0.21 |
| MA (mm) | 51-69 | 19 | 57.2 (54.3-64.1) | -0.2 (-5.8-5.1) | 24 | 55.9 (53.2-64.5) | -0.7 (-7.2-2.1) | 0.71 |
| Ly30 (%) | 0-4 | 19 | 1.0 (0.0-2.7) | 0.0 (-1.6-1.2) | 24 | 2.4 (1.1-6.1) | -1.1 (-3.7-1.1) | 0.20 |
3.3. Treatment Efficacy and Toxicity Measures
There was no significant difference between groups in intraindividual changes in HIV-RNA levels. Few individuals had a viral load > 20 copies/mL. Both CD4 and CD8 counts were higher when receiving ABC compared with TDF, but only significantly so for CD8, with a median change of -30 cells/µL (IQR: -290-160) vs. 120 cells/µL (IQR:-40-230), p=0.04 in the AT and TA group respectively. Changes in CD4/CD8 ratio did not differ significantly between groups. Kidney function and platelet counts were within normal limits and comparable between groups and did not differ during the study period (Supplementary Table 3 and Supplementary Fig. 5).
4. DISCUSSION
To explore the potential biological mechanisms explaining the observed association between ABC and the risk of MI, we assessed changes in Multiplate® and TEG® as well as changes in lipids and a comprehensive range of biomarkers of coagulation, inflammation, platelet reactivity, endothelial disruption and activation and fibrinolysis when switching between ABC and TDF-containing cART regimens. The main findings were unaffected Multiplate® and TEG® (primary and secondary hemostasis parameters); however, there were subtle but significant treatment effects in a procoagulant direction in several biomarkers. These included higher levels of coagulation factors II-VII-X, sCD40L (a biomarker of platelet activation), thrombomodulin (a biomarker of endothelial damage), CD8 (a biomarker of immune activation), and lipids during treatment with ABC compared to TDF.
Platelet aggregation has been assessed in several studies as a proxy for primary hemostasis in PWH. Increased platelet aggregation has been found in treated and naïve
PWH compared to HIV-negative controls [16], and ABC-exposed PWH compared to PWH treated with cART regimens without ABC [17]. Studies where platelets have been directly incubated with NRTIs, including ABC and TDF, have shown no difference in in vitro platelet aggregation upon activation [18]. However, ABC-incubation enhanced platelet granule release when stimulated with collagen, implying that ABC can enhance platelet activation. In a mouse model, ABC enhanced collagen-evoked platelet aggregation in vivo, proposing a mechanistic link between ABC and the risk of MI by enhanced platelet degranulation and interrupted NO-mediated platelet inhibition [18], and believed to be caused by the active anabolite of ABC, carbovir triphosphate [19, 20].
TEG® is a viscoelastic assay to assess secondary hemostasis in whole blood [21].Only very few studies have previously been conducted using TEG® as a functional assessment of coagulopathy in PWH [22-24], and none intend to examine the treatment effects of ABC and TDF. We found no differences between the two treatments.
Several studies have assessed biomarkers to explain the association between ABC exposure and the risk of CVD [10]. Results have been inconsistent partly due to differences in design and methods [10, 25-27]. CD40L is expressed on the surface of activated platelets and immune cells; however, the vast majority of sCD40L originates from activated platelets [28]. sCD40L has previously been shown to be elevated in PWH (naïve and treated) compared to healthy individuals [29], and ABC has been shown to induce platelet activation [11, 17, 19, 30]. In this study, sCD40L levels were higher during treatment with ABC compared to TDF, confirming this observation.
Inconsistently, ABC use has been associated with endothelial dysfunction [10, 26, 31, 32] and increased endothelial inflammation and platelet adherence [33, 34]. Although syndecan-1 and thrombomodulin are excellent proxies for glycocalyx and endothelial cell disruption and/or damage [35-37], they have not previously been examined in relation to ABC. Only thrombomodulin was significantly affected as a sign of endothelial disruption; however, both sE-selectin and syndecan-1 similarly tended to be affected to higher levels during ABC-treatment. Higher levels of coagulation factor II-VII-X during treatment with ABC were a highly significant and novel observation. The biological explanation is unclear and will need to be confirmed in future studies. The APTT is a functional test of the intrinsic and common pathways of coagulation cascade cascades (factors I, II, V, VIII, IX, X, XI, XII). Coagulation factors must be severely reduced before the APTT is affected. Thus, the lack of change in APTT does not exclude that these coagulation factors could be similarly affected.
The CD8-count was higher during treatment with ABC, indicating a higher degree of immune activation. Increased levels of CD8-counts have been associated with all-cause mortality in HIV negative individuals [38] and with non-AIDS-related mortality in PWH [39].
Our finding of increased lipids during ABC treatment compared with TDF-based regimens is well-known and consistent with previously published studies [40, 41].
This is the first study using a cross-over design to approach the topic. The design is the main strength of the study, as it overcomes the ‘confounding bias’ or ‘channeling bias’ that has been a factor of dispute in the debate regarding MI-risk associated with ABC exposure. Additionally, it provides enhanced statistical power and facilitates the detection of even small effects as individuals serve as their controls, thus eliminating interindividual variability and effects of other (perhaps unknown) HIV-associated and unassociated factors.
Furthermore, combining functional hemostasis analyses with a comprehensive panel of biomarkers of both inflammation and coagulation variables provides an opportunity for deep insights into the pathogenesis. With multiple outcome measures and analyses, however, the risk of type I errors exists. This risk can be lowered by, i.e., restricting the interpretation of significance level to a lower p-value. However, as this was an explorative/hypothesis-generating study of possible mechanisms, we did not want to be too restrictive and thereby risk overlooking the true effects (type II error). Furthermore, using biomarkers as proxies for biological processes allows for insights into pathogenesis but may not correlate well with the actual risk of MI.
| Normal Range | AT Group | TA Group | p-values | |||||
|---|---|---|---|---|---|---|---|---|
| N | Baseline (ABC Treatment) | Intra-Individual Change when Switching from ABC to TDF | N | Baseline (TDF Treatment) | Intra-Individual Change when Switching from TDF to ABC | Wilcoxon Rank Sum | ||
| 90%CI | Median (IQR) | Median (IQR) | Median (IQR) | Median (IQR) | ||||
| Biomarkers | ||||||||
| Biomarkers of coagulation | ||||||||
| APTT (s) | 25-37 | 18 | 28.5 (26.0-29.0) | 0.0 (-1.0-1.0) | 21 | 29.0 (26.0-30.0) | 0.0 (-1.0-1.0) | 0.66 |
| Factor II-VII-X (U/L) | 0.70-1.30 | 19 | 1.07 (0.93-1.26) | -0.07 (-0.13-0.04) | 22 | 1.00 (0.84-1.08) | 0.13 (-0.04-0.20) | <0.0001 |
| Platelets (x109/L) | 145-390 | 19 | 212 (187-252) | 4 (-17-22) | 24 | 213 (192-254) | -11 (-23-15) | 0.15 |
| Fibrinogen (mg/dL) | 200-400 | 19 | 276 (248-344) | -7 (-34-31) | 22 | 311 (276-395) | -24 (-65-27) | 0.34 |
| D-dimer (mg FEU/L) | <0.5 | 19 | 0.3 (0.3-0.3) | 0.0 (0.0-0.0) | 22 | 0.3 (0.3-0.3) | 0.0 (0.0-0.0) | 0.56 |
| Antitrombin (%) | 80-120 | 19 | 111 (96-121) | 0 (-5-4) | 22 | 109 (100-120) | -3 (-11-0) | 0.23 |
| Biomarkers of platelet reactivity | ||||||||
| sCD40L (pg/mL) | 19 | 259 (179-453) | -112 (-204- 44) | 24 | 175 (103-735) | 81 (-57-210) | 0.04 | |
| Beta-thromboglobuline (ng/mL) | 19 | 3712 (3068-8257) | -175 (2937-1580) | 24 | 5329 (3458-11258) | -225 (-2969-1847) | 0.86 | |
| PF4 (ng/mL) | 18 | 14.03 (11.32-16.48) | -0.34 (-5.17-2.20) | 23 | 16.68 (11.99-27.36) | -1.22 (-8.30-3.97) | 0.79 | |
| TGF-beta1 (pg/mL) | 19 | 4939 (3635-8143) | -393 (-1224-34) | 23 | 5095 (3437-7042) | 45 (-2125-849) | 0.64 | |
| sGPVI (ng/mL) | 19 | 78.82 (54.10-105.97) | 4.22 (-27.43-11.76) | 24 | 84.15 (58.98-117.03) | 4345 (-22903-29489) | 0.81 | |
| Biomarkers of endothelial disruption and activation | ||||||||
| Se-selectin (ng/mL | 19 | 114.68 (75.45-128.28) | -0.51 (-16.40-6.30) | 24 | 110.57 (92.79-124.70) | 3.28 (-5.44-14.07) | 0.17 | |
| Syndecan-1 (ng/mL) | 19 | 18.11 (9.99-24.99) | -2.12 (-3.35-0.01) | 24 | 15.60 (11.84-23.73) | 2.46 (-3.34-4.68) | 0.15 | |
| Thrombomodulin (ng/mL) | 19 | 3.60 (3.26-3.98) | 0.03 (-0.29-0.29) | 24 | 3.55 (3.30-4.13) | 0.22 (0.04-0.70) | 0.04 | |
| Biomarkers of fibrinolysis | ||||||||
| tPA (ng/mL) | 19 | 2.28 (1.54-3.92) | 0.27 (-0.30-0.68) | 24 | 2.38 (1.77-3.78) | 0.05 (-0.43-0.77) | 0.66 | |
| PAI (ng/mL) | 19 | 53.65 (36.55-84.11) | -8.70 (-28.49--1.24) | 24 | 57.87 (48.31-73.28) | -5.11 (-13.33-3.90) | 0.27 | |
The study's main limitation is the rather low number of study participants, and we acknowledge that the combination of a low number of study participants and comprehensible outcomes increases the risk of type I errors. The decision to end the study early was based on recruitment issues and a high proportion of drop-outs due to side effects, especially in the TA group. This is a reminder that if the patient is content with a cART regimen, the clinician should carefully weigh the possible benefits of treatment alterations before implementing such. Statin use would have been interesting to include in the analysis, as there is increasing awareness of the effects of statins on platelet function and reactivity [42]. However, this information was not possible to retrospectively collect.
CONCLUSION
In conclusion, switching between ABC and TDF did not result in changes in primary and secondary hemostasis, as assessed by Multiplate® and TEG®. However, multiple biomarkers were affected in a pro-coagulant direction during treatment with ABC compared to TDF, with increased levels of coagulation factors (factor II-VII-X) and signs of platelet reactivity (higher sCD40L), endothelial damage (higher thrombomodulin), immune activation (higher CD8-counts), and a less favorable lipid profile. This indicates that the ABC-induced increased risk of MI is multifaceted, including disturbances in both the hemostatic and vascular systems, confirming the current understanding. Despite progress in the field, the perturbed mechanisms are complex and remain incompletely comprehended.
AUTHORS' CONTRIBUTIONS
MH, FR, and JG conceived the study, FR and MH wrote the protocol and received approval from relevant authorities, FR, MH, and JG enrolled the participants, FR and SRO conducted the laboratory analysis, CD, FR, DM, and MH conducted the statistical analysis. CD, DM, and MH wrote the initial draft. All authors contributed to a critical review of the article and approved the final version.
LIST OF ABBREVIATIONS
| MI | = Myocardial Infarction |
| TDF | = Tenofovir Disoproxil |
| NRTI | = Nucleoside Reverse Transcriptase Inhibitor |
| ABC | = Abacavir |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The trial was approved by the Danish Data Protection Agency (30-0981), the Ethical Committee for Biochemical Research in the Capital Region, Denmark (H-2-2013-082) and by the Danish Health and Medicines Authority (2013051722).
HUMAN AND ANIMAL RIGHTS
No animals were used for studies that are the basis of this research. All the humans used were in accordance with the Helsinki Declaration of 1975.
CONSENT FOR PUBLICATION
Informed consent has been obtained from the participants involved.
STANDARDS OF REPORTING
Consort guidelines were followed.
AVAILABILITY OF DATA AND MATERIALS
All data are presented in the manuscript and supplementary material.
FUNDING
The study was supported by grants from Gilead Sciences, the Augustinus Foundation, the Danish AIDS foundation,and the Danielsen Foundation. MH received support from Danish National Research Foundation (#126). The funders had no role in the design, collection, analysis, and interpretation of data in the writing of the manuscript, nor in the decision to submit the manuscript for publication.
CONFLICT OF INTEREST
The results have been presented as a poster at the Nordic HIV and Hepatitis Conference 2015, Stockholm, Sweden. Conflicts of interests: MH: Advisory board: GSK & MSD; travel grant: Gilead, GSK; speaker honoraria: GSK, Gilead, MSD. CD: travel and research grant: Takeda. JG, SRO, DM, FR: no conflicts.
ACKNOWLEDGEMENTS
We thank the participants for their willingness to participate in the study.
SUPPLEMENTARY MATERIALS
Supplementary material is available on the Publisher’s website.

