All published articles of this journal are available on ScienceDirect.

HIV-1 Vaccine Trials: Evolving Concepts and Designs
Authors Info & Affiliations
Abstract
An effective prophylactic HIV-1 vaccine is needed to eradicate the HIV/AIDS pandemic but designing such a vaccine is a challenge. Despite many advances in vaccine technology and approaches to generate both humoral and cellular immune responses, major phase-II and -III vaccine trials against HIV/AIDS have resulted in only moderate successes. The modest achievement of the phase-III RV144 prime-boost trial in Thailand re-emphasized the importance of generating robust humoral and cellular responses against HIV. While antibody-directed approaches are being pursued by some groups, others are attempting to develop vaccines targeting cell-mediated immunity, since evidence show CTLs to be important for the control of HIV replication. Phase-I and -IIa multi-epitope vaccine trials have already been conducted with vaccine immunogens consisting of known CTL epitopes conserved across HIV subtypes, but have so far fallen short of inducing robust and consistent anti-HIV CTL responses. The concepts leading to the development of T-cell epitope-based vaccines, the outcomes of related clinical vaccine trials and efforts to enhance the immunogenicity of cell-mediated approaches are summarized in this review. Moreover, we describe a novel approach based on the identification of SIV and FIV antigens which contain conserved HIV-specific T-cell epitopes and represent an alternative method for developing an effective HIV vaccine against global HIV isolates.
INTRODUCTION
While developing a successful HIV vaccine would arguably be the best method of eradicating the AIDS pandemic, designing such a vaccine is proving to be one of the most significant challenges of the 21st century. Since the discovery of the virus in 1983 [1], HIV-1 has infected more than 60 million individuals and caused greater than 25 million deaths, most of which have occurred in sub-Saharan Africa [2]. Though control of new HIV infections has improved due to major global efforts towards preventive education and access to antiretroviral therapies, new infection rates are still high and the disease remains a significant burden in countries with high HIV-prevalence [3].
Over the past several decades, the primary focus of HIV vaccine research has evolved away from designing a traditional antibody-based vaccine towards a more balanced approach that would activate both humoral and cell-mediated immune (CMI) responses in vaccine-induced protection [4,5]. New delivery systems, novel immunization regimens, and a wide selection of formulations for immunogens and adjuvants have been developed. It is now quite clear that a careful choice of vaccine immunogens capable of affording protection against many variants of HIV is important. Not only must they protect against existing variants, they must also be able to confront a virus with an extraordinarily high mutation rate, a function that drives a continuum of increasing antigenic diversity. This article briefly discusses ongoing research in T-cell-based HIV vaccines and clinical trials that focused on CMI activation. We also describe a new approach to selecting vaccine immunogens based on conserved cytotoxic T lymphocyte (CTL) epitopes.
WORKING TOWARDS AN HIV VACCINE
While some of the most effective vaccines in use today have been based on attenuating the viral pathogen, concerns regarding the potential for reversion to a virulent form of HIV have made the development of an attenuated HIV vaccine unfeasible [6]. Similar concerns related to the safety of inactivated whole viral vaccines have reduced support for a killed HIV vaccine [7]. Challenges to the development of an effective HIV vaccine based on subunits, rather than whole virus include the enormous diversity of viral sequences and the rapid rate of viral evolution that allows HIV to evade protective immune responses [8,9]. Despite recent successes in the identification of broadly cross-reactive neutralizing antibodies (bNAbs) against HIV-1[10], current methods for inducing these antibodies have not been successful [11]. Furthermore, much remains to be learned regarding virus/host interactions and the exact immune correlates of protection; these remain some of the most formidable obstacles to the design of an effective HIV vaccine [12].
Nevertheless, HIV researchers have made substantial progress in understanding the evolution, pathogenesis, and immunological responses to the virus through preclinical and clinical studies in humans and animals [13]. These advances have been achieved by studying a variety of human populations such as vaccinated volunteers, HIV-exposed seronegative individuals (HESN), and different clinical groups of HIV-infected individuals (rapid progressors, elite controllers, and long-term survivors or non-progressors, see below). Experimental vaccine studies have also been conducted in animals. For example, immune responses to simian immunodeficiency virus (SIV) and recombinant SHIV have been extensively studied in macaques [14-16]. A viral infection that resembles HIV, equine infectious anemia virus (EIAV) has been studied in horses [17], and similarly, caprine arthritis encephalitis virus (CAEV) has been the subject of extensive research in goats [18,19]. Our group has focused on elucidating immune responses to feline immunodeficiency virus (FIV) in cats [20,21]. More recently, HIV viral vectors have been used to study cell-mediated immune responses in rodent models, and the hu-SCID mouse has been considered as a potential model for vaccine development [22-25] (Table 1). New approaches and technologies utilized in HIV research are advancing science in a multidisciplinary way to face the ongoing scientific challenges of developing both an HIV vaccine and immunotherapeutic strategies.
Animal Models in HIV-1 Research
| Animal Models | Virus | Major Cell Types Infected | Major Clinical Disorders | Refs.b |
|---|---|---|---|---|
| Macaques | SIV/SHIV | Macrophages, CD4+ T cells | Immune deficiency, AIDS-like | [14-16] |
| Cats | FIV | Macrophages, CD4+ T cells, CD8+ T cells | Immune deficiency, AIDS-like | [20,21] |
| Horses | EIAV | Macrophages | Autoimmune hemolytic anemia | [17] |
| Goats | CAEV | Macrophages | Arthritis, Encephalopathy | [18,19] |
| Rodent models (e.g., hNOG) a | HIV-1 (vectors) | Macrophage-like cells, CD4+ T cells | CD4+ T-cell loss | [22-25] |
a Humanized NOD/SCID/IL2Rnull (hNOG): severely immunodeficient mice that can easily engraft human cells.
b Reference number (Refs.).
Immunogens Used in Major Clinical Vaccine Trials
| Vaccine Trial Phase (Vaccine Name) | Trial Location | HIV-1 Immunogens (Vaccine type) | Trial Outcome | Refs.a |
|---|---|---|---|---|
|
|
||||
| VaxGen 004 | North America | Subtype B gp120 (subunit proteins) | No efficacy | [27] |
| Phase III | ||||
| (AIDSVAX B/B) | ||||
|
|
||||
| VaxGen 003 | Thailand | Subtypes B & E gp120 (subunit proteins) | No efficacy | [26, 28] |
| Phase III | ||||
| (AIDSVAX B/E) | ||||
|
|
||||
| Step HVTN 502 | Americas | Subtype B Ad5-gag/pol/nef (Ad5 vector ) | No efficacy (enhancement of infection) | [29,30] |
| Phase IIb | ||||
| (MRKAd5 HIV-1) | ||||
|
|
||||
| RV144 | ||||
| Phase III | Thailand | Prime - subtype B and A/E | Some efficacy | [34] |
| (ALVAC-HIV-1 | ALVAC-HIV-gag-pr-gp41-gp120 (canarypox vector) | [General population (31.2%) | ||
| AIDSVAX B/E) | Boost - Subtypes B & E gp120 (subunit proteins) | [High risk groups (3.7%)] | ||
a References (Refs.).
A T-Cell Vaccine Based on Better Disease Outcome in HIV+ Individuals
| Characteristics of CD4+ and/or CD8+ T Cells | Refs.b |
|---|---|
| Have a large T-cell receptor repertoire diversity | [99] |
| Express markers of central and effector memory functions | [104,105] |
| Be able to destroy HIV-1 infected cells or suppress HIV-1 replication ex vivo | [102,103] |
| Be able to proliferate upon HIV antigenic stimulation | [101] |
| Be polyfunctional in cytokine production upon HIV antigenic stimulation | [100] |
| Be specific for several Gag epitopes that are restricted by HLAs associated with viral control and conserved across many subtypes | [107-111] |
| Not express markers of T-cell exhaustion (e.g., PD-1, LAG-3, Tim-3, and CTLA-4)a | [106,112-114] |
a Programmed cell death 1 (PD-1); lymphocyte activation gene 3 (LAG-3); cytotoxic T lymphocyte-associated antigen 4 (CTLA-4); T cell immunoglobulin mucin-3 (Tim-3).
b References (Refs.).
Phase I and IIa Clinical Trials of HIV CTL Multi-Epitope Vaccine
| Triala | Site (# of Subjects Enrolled in the Study)b | Vaccine Type (Regimen)c | Dose/Route DNA (mg), MVA (p.f.u.)d | HIV Antigens (# of CTL Epitopes)e | HIV Subtypes Involvedf | HLA Super-Type of CTL Epitopes | Epitope Selection Method | (%) IFNγg Responders | Refs.h |
|---|---|---|---|---|---|---|---|---|---|
| IAVI-001 | UK (18) | DNA (d: 0,21) | 0.1 or .05 mg/i.m. | p24/p17 gene [contains TH epitopes] + 24 CTL epitopes [p24(6), pol(6), nef(8), Env (4)] | A* [A, B, C, D, E, F, G, H]j | A2, A3, A24, B7, B8, B27, B44 | Most common HIV subtype in Kenya Conserved epitopes | 78% | [129, 130] |
| IAVI-002 | Kenya (18) | DNA (d: 0,21) | 0, 0.1 or 0.5 mg/i.m. | 15% | [131] | ||||
| IAVI-003 | UK (8) | MVA (d: 0,21) | 5x107 p.f.u./i.d. | 78% | [129] | ||||
| IAVI-004 | Kenya (18) | MVA (mo: 0,1 or 0) | 0 or 5x107 p.f.u./i.d. | 25% | [131] | ||||
| IAVI-011 | Switzerland, UK, SA (81) | MVA (mo: 0,2) | 0, 5x106, 5x107 or 2.5x108 p.f.u./i.d., i.m. or s.c. | 6% | [132] | ||||
| IAVI-005 | UK (9) | p-DNA (d: 0,21)i b-MVA | 0.1 or 0.5 mg/i.m. 5x107 p.f.u./i.d. | 89% | [129] | ||||
| IAVI-006 | UK (119) | p-DNA (mo: 0) b-MVA (mo: 2, 3 or 5,6) | 0, 0.5 or 2 mg/i.m. 0 or 5x107 p.f.u./i.d. | 12% | [132] | ||||
| IAVI-008 | Kenya (10) | p-DNA (d: 0,21) b-MVA (mo: 9, 10) | 0.5 or 1 mg/i.m. 5x107 p.f.u./i.d. | 10% | [131] | ||||
| IAVI-009 | Uganda (50) | p-DNA(mo: 0,1 or 0) b-MVA (mo: 5,8) | 0 or 0.5 mg 1x or 2x/i.m. 0 or 5x107 p.f.u./i.d. | 15% | [131] | ||||
| IAVI-010 | Kenya, UK (114) | p-DNA (mo: 0,1) b-MVA (mo: 5,8) | 0.5 mg/i.m. 0, 5x106, 5x107 or 2.5x108 p.f.u./i.d. | 3% | [132] | ||||
| IAVI-016 | UK (24) | p-DNA (mo: 0, 1) b-MVA (mo: 2 or 0,1) | 0 or 4 mg/i.m. 0 or 2.5x108 p.f.u./i.d. | 50% | [133] | ||||
| HVTN-048 | USA, Bostwana (36) | DNA (mo: 0,1,3,6) | 0.5 mg 4x/i.m. | 21 CTL epitopes [Gag (4), Pol (8), Vpr (1), Nef (2), Rev (1), Env (5)] + TH epitope (1 pan-DR) | A, B, C, D, AE, AG | A2, A3, B7 | Conserved Epitopes HLA coverage | 0% | [134] |
| 2 mg 4x/i.m. | 0% | ||||||||
| 4 mg 4x/i.m. | 13% | ||||||||
| HVTN-056 | USA (40) | MEP [peptides + adjuvant] (mo: 0,1,3) | 1 mg MEP + 50 µg adjuvant 3x/i.m. | 4 peptides (55 CTL epitopes): Env-TH/ Gag-CTL (5), Gag-TH/ Gag-CTL (19), Env-TH/ Nef-CTL (15), Env-TH/ Gag-CTL (16) | B* | A1, A2, A3, A24, B7, B8, B27, B58, B62 | Epitope clustering on LANL | 13% | [135] |
| USA (40) | MEP [peptides + adjuvant + GM-CSF ] (mo: 0,1,3) | 1 mg MEP/50 µg adjuvant + 50 ug GM-CSF 3x/i.m. | 3% | ||||||
|
ANRS VAC18 |
France (99) | Lipopeptides (mo: 0, 1, 3, 6) | 50 µg 4x/i.m. | 5 lipopeptides (77 CTL epitopes, containing 7 TH epitopes): [Gag1 (9), Gag2 (21), Nef1 (16), Nef2 (21), Pol (10)] | A1, A2, A3, A24, B7, B8, B27, B58, B62 | Conserved regions | 71%k | [136] | |
| 150 µg 4x/i.m. | 60%k | ||||||||
| 500 µg 4x/i.m. | 70%k |
a All trials are phase I clinical trials except for the bolded trial numbers which are phase IIa (with subjects not at risks of HIV infection); International AIDS Vaccine Initiative (IAVI); HIV Vaccine Trials Network (HVTN); Agence National de Recherche sur le SIDA (ANRS).
b United Kingdom (UK); South Africa (SA); United States of America (USA).
c Prime (p); boost (b); day (d); month (mo); modified vaccinia Ankara (MVA); multi-epitope peptide (MEP); granulocyte macrophage colony stimulating factor (GM-CSF).
i Nine of the 18 volunteers from IAVI-001 who were primed with HIVA-DNA agreed to receive a boost 9-14 months later.
d Intramuscular immunization (i.m.); intradermal immunization (i.d.); subcutaneous immunization (s.c.).
e MHC class I molecules can accommodate CTL epitopes of 8 to 11 aa in length [137]. The p24/p17 represents 73% of the Gag and contains both CTL and T-helper epitopes. The pan-DR T-helper epitope is a 13-mer that binds to all common HLA-DR alleles. Each of the four peptides in the MEP vaccine is made up of both TH and CTL epitopes; T helper (TH).
f The HIV subtypes used in the vaccine.
* Consensus sequence.
j The CTL epitopes are present in 50-90% of HIV isolates from the different subtypes.
g Percentage of vaccinees with detected IFNγ ELISpot responses to the CTL epitopes. The responses were detected at different time points, before or after the end of the immunization schedule for the IAVI studies; after the last immunization for HVTN 064; and after the 2nd or 3rd vaccination (single time point) for HVTN 056.
k Cultured ELISpot assay results.
h References (Refs.).
HIV-1/FIV Proteins
a The average Shannon entropy score is the average value of variability of a given protein at each aa position, calculated by using many aligned sequences. The approximate values shown are derived from the figure of HIV-1 (group M) protein variability from Yusim et al. [161], where the proteins are presented from lowest to highest variability. Lower scores represent lower variability and therefore higher aa conservation.
b The percentage of aa identity and homology between HIV and FIV proteins are shown, with the three most conserved HIV and FIV proteins with *.
c References (Refs.).
CTL-Epitope Prediction Tools
| Name | Website | Developer | Refs.a |
|---|---|---|---|
| CTLPred | http://www.imtech.res.in/raghava/ctlpred/ | India | [168] |
| NetCTL | http://www.cbs.dtu.dk/services/NetCTL/ | Denmark | [169] |
| NetCTLpan | http://www.cbs.dtu.dk/services/NetCTLpan/ | Denmark | [170] |
a References (Refs.).
Best Defined CTL Epitopes on HIV Integrasea
| Epitope | Position on HXB2 | HLA | |
|---|---|---|---|
| 1 | LPPIVAKEI | 28-36 | B42 |
| 2 | THLEGKIIL | 66-74 | B*1510 |
| 3 | STTVKAACWW | 123-132 | B57 |
| 4 | IQQEFGIPY | 135-143 | B*1503 |
| 5 | VRDQAEHL | 165-172 | Cw18 |
| 6 | KTAVQMAVF | 173-181 | B*5701 |
| 7 | AVFIHNFKRK | 179-188 | A*0301, A*1101 |
| 8 | FKRKGGIGGY | 185-194 | B*1503 |
| 9 | KRKGGIGGY | 186-194 | B*2705 |
| 10 | IIATDIQTK | 203-211 | A*1101 |
| 11 | KIQNFRVYY | 219-227 | A*3002 |
| 12 | VPRRKAKII | 260-268 | B42 |
| 13 | RKAKIIRDY | 263-271 | B*1503 |
a Adapted from LANL (http://www.hiv.lanl.gov/content/immunology/tables/optimal_ctl_summary.html) which was last updated on 2009-08-31. The best defined CTL epitopes or “A list” represent the epitopes whose specific HLA class I allele has been demonstrated with strong certainty and are judged to be at their optimal length.
HIV-1 Integrase CTL Epitopes and Direct FIV Counterparts
| Allele (Supertype) | Virus | Epitopesa | Iden.b | Hom.b | IEDB Prediction: Binding Allele (nM Value)c | Supertype (Total # of Binding Alleles) | NetCTL Supertype |
|---|---|---|---|---|---|---|---|
| B*1510 (B39) | HIV | THLEGKIIL | B*3901(9); B*1501(425) | B39(2) | B39 | ||
| SIV | THLEGKIII | 78 | 100 | B*3901(44) | B39(1) | B39 | |
| FIV | THFNGKIII | 56 | 78 | B*3901(64); B*1501(373) | B39(2) | B39 | |
| A*0301 (A3) A*1101 (A3) |
HIV | MAVFIHNFK | A*0301 (363); A*1101 (20) | A3(5); A1(1) | A3 | ||
| SIV | MAVHCMNFK | 67 | 67 | A*0301(174); A*1101(25) | A3(4); A1(1) | A3 | |
| FIV | LALYCLNFK | 44 | 78 | A*3001(113); A*1101(55) | A3(3); A1(1) | A3 | |
| B42 (B7) | HIV | VPRRKAKII | B*0702(43); B*0801(53) | B7(1); B8(1) | B7; B8 | ||
| SIV | VPRRKAKII | 100 | 100 | B*0702(43); B*0801(53) | B7(1); B8(1) | B7; B8 | |
| FIV | VPRRHIRRV | 44 | 67 | B*0702(20); B*0801(124) | B7(1); B8(1) | B7; B8 | |
| A*1101 (A3) | HIV | IIATDIQTK | A*1101(404); A*6801(204) | A3(3) | A3 | ||
| SIV | ILATDIQTT | 78 | 89 | A*0250(10) | A2(5) | None | |
| FIV | QESLRIQDY | 22 | 33 | B*4402(88) | B44(3) | None | |
| FIV | IVAEEIKRKd | 44 | 78 | A*1101(338); A*6801(206) | A3(2) | A3 |
a The HIV epitope sequences are from the LANL list of the best defined CTL epitopes for HIV integrase. The SIV counterpart sequences are derived from LANL SIVmm239 and the FIV counterpart sequences are derived from GenBank (ABD16378) after aa alignment with HXB2 sequence.
b The identity (iden.) and homology (hom.) values were obtained using EMBOSS Stretcher - Pairwise Sequence Alignment (http://www.ebi.ac.uk/Tools/psa/emboss_stretcher/).
c MHC binding for HIV, SIV and FIV counterpart epitopes were predicted using the Immune Epitope Database (IEDB) MHC class I binding prediction tool (http://tools.immuneepitope.org/analyze/html/mhc_binding.html).
The matching binding alleles are shown along with their binding affinity values (nM) which are derived from the Artificial Neural Network (ANN) analysis, where lower values represent higher binding affinity and potential for CD8+ T-cell activity. The total numbers of binding alleles with affinity below 500 nM are shown in parenthesis next to the supertypes.
d HIV epitope with non-matching SIV and FIV (direct counterparts) is in italics and the bolded FIV epitope is an indirect counterpart with matching alleles to the HIV epitope.
MAJOR PHASE-II AND PHASE-III CLINICAL VACCINE TRIALS
VAX004, which was initiated in 1998, was the first phase-III human trials to be conducted. The immunogens for this study were recombinant surface envelope glycoproteins (gp120) from two HIV-1 subtype-B viruses (AIDSVAX B/B) [26] (Table 2). The second phase-III trial VAX003, initiated in 1999, used HIV-1 subtype-B and subtype-E glycoproteins (AIDSVAX B/E) as immunogens [27,28]. Both of these vaccines were designed to stimulate humoral immune responses but failed to produce potent neutralizing antibodies (NAbs) against clinical HIV isolates and, consequently, afforded no protective efficacy. Following these two unsuccessful attempts, researchers focused on means of inducing CTL responses to confer protection. These efforts culminated with the STEP vaccine trial (Table 2). The vaccine used in this study was designed to induce strong CMI responses [29]. It consisted of an adenovirus serotype-5 (Ad5) vectored HIV-1 gag/pol/nef from three subtype B strains (CAM-1 gag, JRFL pol (RT & IN), IIIB nef) [30].
Initiated in 2004, the phase IIb trial was terminated before completion due to higher HIV infection rates among vaccine recipients than among placebo recipients [29,30], possibly explained by the Ad5 sero-positivity status and lack of circumcision of the men who became infected [29-31]. This increased risk of HIV infection was shown to fade over time in a four-year follow-up study [32]. Outside of the context of vaccination, Ad5-neutralizing antibodies did not increase the risk of HIV infection in a high risk population, even after adjusting for their circumcision status [33].
RV144, which was initiated in 2004, is the most recently completed phase-III vaccine trial. The vaccine consisted of a canarypox virus-vectored HIV-1 gag/pol/env for priming and the AIDSVAX B/E for boosting. This vaccine was intended to induce both humoral and CMI responses and to cover a wider range of potential challenge strains. The vector expressed both the gag-pr-gp41 genes from subtype-B LAI strain and a gp120 from a circulating recombinant isolate called CRF01_AE. The phase III trial, which involved more than 16,000 subjects, demonstrated only modest overall efficacy (31.2%), and a very minimal efficacy (3.7%) in the high risk group [34]. Detectable antibodies and lymphocyte proliferation responses were observed during the phase II trial, where the majority of vaccine recipients also demonstrated antibody-dependent cell mediated cytotoxicity (ADCC) responses [35,36]. Following phase III, IgG antibodies binding to the V1/V2 variable loops of Env were found to correlate with protection, while serum IgA to Env correlated with increased risk of infection without causing an enhancement of infection rates [37,38].
In summary, during the past three decades of HIV vaccine research, only three candidate vaccines have completed phase-III clinical trials [39], reflecting the need for additional research and development of innovative approaches. Furthermore, in these phase-III trials, only the HIV-1 subtype B (three strains) and a recombined HIV-1 subtype A/E (gp120 from CRF01_AE) were used as vaccine immunogens. Although these strains and subtypes reflect the circulating forms of HIV that are more prevalent in the geographical location of the trials, they may not contain optimal immunogens needed for a vaccine effective against a wider range of HIV isolates. The relative success of the RV144 trial suggests that an HIV vaccine may be possible using current technologies. In the next few sections of this review, we address aspects of HIV vaccine development and propose a path forward that may improve on this modest successs.
POTENTIAL CORRELATES OF PROTECTION
An immune correlate of protection can be defined as an immune response that is normally associated with protection from infection or disease and for which a measurable threshold can be defined [40]. In the case of HIV/AIDS, the study of HIV-exposed seronegative individuals (HESN) and HIV-positive (HIV⁺) elite controllers (ECs) has provided significant insights into the immune correlates of protection from infection and disease. Furthermore, studies of the RV144 trial have made contributions by defining more precise vaccine-induced immune correlates of protection in humans. When fully identified these correlates should provide a turning point for advancing HIV vaccine development [41].
HIV-exposed seronegative is a term that refers to cohorts of commercial sex-workers, hemophiliacs, discordant couples, intravenous drug-users, and mother-to-child cases, who, despite having high risks or documented exposures to HIV, have remained uninfected [42]. While a range of factors can potentially explain how HESN remain uninfected, a range of conditions including genetic mutations, T-cell specific HIV responses, CD8+ cell antiviral activities, components of innate immunity, or intracellular intrinsic factors have been considered, but conclusions regarding the causes of these conditions are not evident. Efforts are currently underway to standardize the above studies and assays and explore other avenues [42,43].
Elite controllers have been a major focus of many studies. They are HIV⁺ individuals who are able to maintain undetectable viral loads (<75 RNA copies/mL) for many years without the use of antiretroviral therapy [44]. These individuals are relatively rare, representing less than 1% of all HIV infections [45]. The majority of ECs exhibit even slower rates of CD4⁺ T-cell decrease and slower disease progression than regular long-term non-progressors [46-48]. CD8⁺ T cells play a central role in controlling HIV replication in ECs [49-51]. Selected genetic factors have also been associated with an elite control of the viral infection, including the CCR5 co-receptor 32 base-pair deletion or the killer cell immunoglobulin-like receptors of NK cells [48,52]. HLA-B57 and -B27 are also over-represented in EC populations and have been correlated with a lower risk of disease, as opposed to certain HLA-B35 alleles which are associated with a higher risk for disease progression [52,53]. Recent results of the International Controller Study, in addition to confirming previous findings, showed that the major genetic determinants associated with control of HIV infection are located in the peptide binding groove of the MHC class I molecule (primarily HLA-B), and also implicated HLA-C and other polymorphisms linked to NK cells [54]. Though these genetic factors can explain about a fifth of the variability in virus control, the question remains to be answered on whether the induction of the same cellular responses can be elicited in individuals who carry different HLA alleles [55]. Moreover, HLA-B57 and -B27 are associated with rapid disease progression in some individuals [56].
CMI responses have been associated with the initial control of HIV infection and reflected a non-cytotoxic response against the virus [57]. The important role of the CD8⁺ CTLs in the control of HIV disease has subsequently been reported [58-61]. In addition, CD4⁺ CTLs play an important role early in the control of other viral infections [62,63]; these have also been shown to be relevant in the case of acute HIV infections [64]. Furthermore, a study of rabies vaccination has demonstrated that NK cells can serve as effectors in acquired or adaptive immunity as a result of CD4⁺ T-cell activation [65]. Similarly, NK cells have been described in other viral infections to possess key adaptive immune features such as memory response and antigen-specific proliferation [66-68]. Consequently their involvement in vaccine design is becoming better appreciated. With respect to HIV, NK cells have also been associated with the prevention of mother-to-child and heterosexual HIV transmission [69,70]. Some studies suggest that they may play a role in the control of HIV replication by responding to peptides [69,71], and may be influenced by the peptide/HLA-B complex via the KIR3DL1 and KIR3DS1 molecules [55].
The role of antiviral antibodies in HIV infection has been demonstrated by passive transfer studies with NAbs, which showed that anti-HIV NAbs prevented SHIV infection in macaques [72] and by anti-HIV NAbs that delayed virus rebound after interruption of antiretroviral therapy (ART) in humans [73]. However, ECs showed no significant difference in NAbs when compared to patients on ART [74]. In addition only low levels of bNAbs have been observed in the majority of ECs, a population that is heterogeneous in terms of immunological characteristics [75,76]. Nevertheless, while ECs can have strong CD8⁺ cell-mediated anti-HIV responses, their antiviral antibody responses may still play a role in the control of HIV replication [77,78].
PROPOSED METHODS FOR EFFECTIVE PROTECTION AGAINST HIV: THE T CELL-BASED VACCINE
A successful HIV vaccine that is able to control the AIDS pandemic could have a variety of positive outcomes, such as inducing a “sterilizing” immunity; a transient infection that is later cleared; or a long-term control of the virus without disease manifestations and without further HIV transmission from the vaccinated individuals. The more conservative and accepted view is that both humoral and CMI responses will be important in such a vaccine. Based on the lessons learned from successful vaccines against other organisms, one correlate of protection has been a defined antibody titer directed against one of the key antigens of those organisms [4]. This correlate is usually derived from the assessment of immune responses in recovered patients who are immune to subsequent infections and serves as a benchmark titer for the development of an effective vaccine. With those conventional vaccines and immune individuals, the induction of specific antibodies is the probable cause of protection against future infections [79]. Although CD4⁺ T cells participate in the B-cell response and memory, T cells in general are thought to play a more dominant role in the control of established infections rather than in prophylaxis [4].
Induction of antibody responses may be critical to developing an effective vaccine, but it has been difficult to identify the proper immunogen. Due to the mounting difficulties associated with making a conventional antibody-based HIV vaccine [80], some researchers have returned to exploring T-cell-based approaches. Designing T-cell mediated vaccines requires careful planning so as to generate immune responses in the context of most human leukocyte antigens (HLA). HLA molecules differ from one individual to another and their prevalence also varies in different populations, a factor that has been a cause for concern about T cell-mediated vaccines [81]. However, HLA class I and class II alleles can be grouped into supertypes which are clusters of HLA molecules sharing overlapping peptide binding specificity [82,83]; vaccines containing epitopes that can be presented by HLA supertypes have a better chance of global success. Three HLA class I supertypes (HLA-A2, A3, and B7) have the potential to cover 80-90% of the major ethnicities [81]. Studies have shown that not all CTL responses are equally effective. For example, inclusion of epitopes presented by some B7 supertype alleles targeting the same epitopes may exert differential pressure on the virus [84]. Nonetheless, a T-cell-based vaccine using the supertype concept may be able to induce similar qualitative T-cell responses against the virus among individuals with diverse HLA alleles.
Most HIV infections occur via sexual transmission at the mucosa [85], which is a replication site and a reservoir for the virus [86,87]. Therefore, if CMI is important, an effective vaccine should induce protective responses at the mucosal surface where the majority of the body’s T cells reside [88]. Some researchers have reported that CD8⁺ CTLs from the blood and mucosa mirror each other [89], with some of them originating from the same clones or sharing the same HIV-specific epitopes and MHC restrictions [90]. In chronically infected individuals, antigens recognized by mucosal T cells are also recognized by peripheral blood mononuclear cells (PBMC) with a few exceptions [91,92]. As a result, the study of these individuals may help define some important correlates of protection that are relevant against HIV at the mucosa.
Findings from non-human primate studies have shown that the picture of the systemic immunity obtained from the study of PBMCs may only be a snapshot of the immunity present at the mucosa [93,94]. Notably, in a study recapitulating human HESN where macaques were intentionally exposed to low doses of SIV rectally, potent virus-specific CTL responses were induced and readily measured in the animals’ intestinal lamina propria mononuclear cells in the absence of detectable virus and virus-specific humoral and T-cell responses in the blood. Importantly, when these responses were present, they were universally associated with protection from mucosal challenge with much higher doses of virus [95]. One caveat still remains at which site immune correlates of virological control can be identified. In a recent macaque therapeutic trial assessing the effect of a particle mediated DNA vaccine administered during antiretroviral therapy, it was shown that the breadth of epitope recognition observed in the mucosa, but not the blood, correlated with control of virus rebound when therapy was stopped [96]. Such findings emphasize the need for parallel vaccine trials in macaques and humans to better understand the role of mucosal T-cell responses in protection and help define surrogate correlates of protection in the blood of humans.
Based on the studies of HIV⁺ individuals, a protective T-cell-based HIV vaccine should have a set of defined characteristics [40] (Table 3). The vaccine should stimulate both CD4+ and/or CD8+ T cells [97,98], which should have a large T-cell receptor repertoire (TCR) diversity [99], an ability to secrete multiple cytokines [100], proliferate [101], destroy HIV infected cells or at least suppress viral replication ex vivo [102,103]. These cells should express markers for central or effector memory functions [104,105], and should not express markers of T-cell exhaustion [106]. They should be specific for several Gag epitopes [107,108] restricted by HLAs associated with viral control [109] and conserved across subtypes [110], particularly since mutations in Gag would impair the fitness of the virus [111]. This extensive list of attributes are only associated with cases of better disease outcome and highlights the difficulties of making a vaccine based on T-cell immunity.
Lessons learned from the major trials can also help design better T-cell vaccines. The possible explanations for the lack of efficacy in the STEP study are important factors to consider in the context of the anti-HIV immunity generated. Pre-existing Ad5-specific cellular responses conserved among many adenoviruses were shown to mitigate T-cell responses against the HIV-1 insert [115]. Individuals with lower Ad5-neutralizing antibodies before vaccination generally had higher T-cell responses to one or more HIV-1 proteins [30]. Furthermore, the vaccine induced a lower frequency of the T-cell responses against the conserved HIV epitopes than against the variable epitopes [116]. The few favorable notes from the STEP trial are also factors to consider. Lower HIV-1 RNA levels were observed in vaccine recipients carrying HLA alleles associated with control of the virus than for the matched-placebo recipients [117]. The vaccine used in the STEP trial (mainly a population of men who have sex with men) was simultaneously tested in a heterosexual cohort in South Africa. In this study, the increased risk of HIV-1 infection was not observed, even at higher titers of pre-existing Ad5-neutralizing antibodies in men. In contrast, favorable CD4⁺ T-cell counts were observed at 2 and 3 months after infection, and hints of lower viral set points, were observed in women [118]. These findings indicate that a careful choice of vaccine vector, with focus on conserved HIV-1 epitopes, and efforts to increase the breath and magnitude of the responses against HIV are likely to improve future T-cell vaccines [116].
The Env-antigen in an Ad26/MVA vector vaccination has been shown to be essential for protection against a heterologous neutralization-resistant SIV challenge in primates where protection correlated with Env-binding antibodies [119]. A rhesus cytomegalovirus SIV vectored (RhCMV) vaccine without Env was able to control a pathogenic SIV below detectable levels for more than a year. This study also shows that a replicative vector like RhCMV can induce SIV-specific effector-memory CD8⁺ T cells (TEM) important for early protection, while a non-replicative vector like the Ad5 induces central memory T cells (TCM), which is useful in the control of viremia [120]. In the human RV144 vaccine trial where IgG antibodies to Env correlated with protection, the vaccine induced stronger humoral than cellular immune responses, along with a predominance of CD4⁺ T cells [34-36] including polyfunctional CD4⁺ CTL against a V2-Env region [121]. Therefore, a vaccine design optimized for also inducing CD8⁺ T-cell responses in addition to the CD4⁺ T-cell and humoral responses is likely to improve on the current success.
MULTI-EPITOPE VACCINES TO INDUCE T-CELL IMMUNITY AGAINST HIV
A multi-epitope vaccine can be defined as a vaccine construct containing a string of individual epitopes, designed to elicit targeted immune responses to the selected epitopes, which can be the most conserved or the most recognized by common HLAs [41,122,123]. Computational methods for minimizing the generation of junctional immunogenicity in between epitopes have been used to construct epitope strings [123]. Multi-epitope vaccines have been shown to be protective in animal models and in mice against lethal challenge doses of vaccinia virus [124-126]. There are relatively few multi-epitope CTL vaccines that have undergone phase I and II clinical trials against HIV. These vaccines were generally safe, but poorly immunogenic (Table 4), most likely due to the vaccine vehicle and the limited number of epitopes contained in the vaccine. The traditional interferon-γ (IFNγ) ELISpot assay is an assay that is widely used in vaccine trials because of its relative efficiency, and has been the main method of assessing HIV-specific T-cell immunogenicity in the majority of the trials described in Table 4.
A multi-epitope vaccine consisting of a concensus subtype-A Gag-p24/p17 linked to a stringed gene of CTL epitopes (termed HIVA) has been evaluated in a number of different trials from the International AIDS Vaccine Initiative (IAVI) including prime-boost designs, as opposed to the HIV Vaccine Trials Network (HVTN) and the Agence National de Recherche sur le SIDA (ANRS) trials described in Table 4 [127,128]. The initial phase I studies of the HIVA vaccine were conducted in the UK (IAVI-001, -003, and - 005) [129,130], in parallel with sister studies in Kenya (IAVI-002, -004, and -008) and in Uganda (IAVI009, phase I/IIa) [131]. Contrasting results were associated with these trials. Even though IFNγ responses were induced, they were transient, and the vaccine induced responses in a higher percentage of vaccinees in the UK (78%, 88%, and 89%) than in Kenya (15%, 33%, and 10%) or Uganda (13%). Subsequent phase I/IIa studies of IAVI-006 and -010 [132] that were conducted with a larger number of volunteers also showed poor vaccine immunogenicity, two weeks after the last MVA vaccination. These results could have been due to low antigen doses or to the timing for testing the immunogenicity, as the responses were later observed to peak one week after the MVA vaccination, and to diminish by the second week.
In the IAVI-016 trial, four of eight (50%) DNA-MVA vaccine recipients showed positive IFNγ and five of eight (63%) recipients demonstrated proliferation responses, both peaking one week after vaccination [133]. This trial also used a cultured IFNγ-ELISpot in which PBMCs were stimulated with the target antigens and cultured for 11 to 13 days prior to being tested for immune responses to epitopes in the ELISpot assay. This procedure improved detection of immunogenicity. A similar approach was recently used in a malaria vaccine trial, in which IFNγ levels in the cultured assay correlated with vaccine-induced protection [138]. Subsequently, in the IAVI-016 trial, eight of eight (100%) volunteers who received the prime-boost vaccination and four of eight (50%) who received two doses of the MVA vaccine showed CD4⁺ T-cell specific IFNγ responses. In addition five of the 12 (42%) responders also had CD8⁺ T-cell specific IFNγ responses to the string of CTL epitopes.
Another trial (HVTN-048) consisted of four immunizations of a DNA vaccine (expressing a 277-aa antigen) containing 21 CTL epitopes. Immune responses to the epitopes were monitored up to 18 months after the last immunization. Only 1 of 8 (13%) recipients of the highest dose of vaccine (4 mg) showed a positive IFNγ-ELISpot response two weeks after the last immunization. Notably, only three CD8⁺ CTL responses were transiently detected using the chromium release assay [135]. In the HVTN-056 trial, the immunogen was a multi-epitope peptide (MEP) vaccine consisting of four peptides (27-47 aa) supplemented with an adjuvant in the presence or absence of granulocyte/macrophage colony-stimulating factor (GM-CSF). Initially intended as a priming agent for a prime-boost vaccine regimen, the vaccine was only weakly immunogenic, as only 6 of 80 vaccinees showed a transient IFNγ response after the second or third vaccination. The use of GM-CSF in the formulation did not improve the immunogenicity of the vaccine [135].
The ANRS VAC18 was a phase II trial consisting of four intramuscular injections of a lipopeptide vaccine. This vaccine contained an equal weight mixture of 5 HIV-1 peptides (19-32 aa) linked to a palmitic acid for easy uptake by antigen-presenting cells. A cultured-IFNγ assay was used to measure immunogenicity. Regardless of the vaccine dose administered (50, 150, or 500 μg) IFNγ responses to the CTL epitopes were repeatedly detected in the majority of vaccine recipients for 12 months after the last immunization. Proliferative responses were also detected to the CD4⁺ T-cell epitopes [136]. This lipopeptide vaccine was also used in a post-infection immunization study, where it induced new CD4⁺ T-cell proliferation and CD8⁺ T-cell IFNγ responses in HIV-1 infected subjects [139].
The vaccines described above and in Table 4 used only a modest number of selected CTL epitopes (21-77). In general, these vaccines were unable to induce the desired breadth of CD8⁺ CTL responses. This may be related to the HLA-restriction of the study subjects, even though the multi-epitope vaccine HVTN-048 and HVTN-056 trials required immunized subjects to carry at least one of the HLA alleles targeted by the vaccine. In the IAVI-009 trial, the vaccine was expected to induce at least two to three CTL-epitope responses per individual. An important concern related to these vaccines is whether the numbers of epitopes used will suffice to induce an effective protection against a wide range of global HIV isolates. The impact of immunizing study subjects against variant viral isolates cannot be tested until a sufficient level of immunogenicity is obtained for the vaccine epitopes in the target populations.
A different clinical trial (HVTN-064) used a multi-epitope vaccine consisting of 18 T-helper epitopes formulated in aluminum hydroxide. This T-helper vaccine was immunogenic in 32 of 47 individuals (68%) by intracellular cytokine staining, and the CD4⁺ T-cell responses observed were polyfunctional. However when administered simultaneously with the same DNA vaccine used in the HVTN-048 trial, this vaccine was unable to improve the immunogenicity towards the CTL epitopes. This may be related to the initial inability of the DNA vaccine alone to induce sufficient CTL responses [140].
The HIVA prime-boost vaccination approach has also been used as a post-infection immunization strategy in phase I trials for HIV-1 infected subjects. Despite initial concerns that immunization might boost viral titers, the vaccine was safe and effectively induced an expansion of CD4⁺ and CD8⁺ T cells specific for vaccine epitopes without viral load rebound [141,142]. Another study utilized the ELISpot assay to evaluate IFNγ secretion in vitro by CD8⁺ T cells following stimulation with either the MVA.HIVA vector or HIV-1 peptides used in the construct. The PBMCs obtained from 81 HESN infants show that MVA.HIVA induced IFNγ responses in 52% (42/81), but only 15% (12/81) of these had IFNγ responses to the individual HIV-1 peptides [143]. An additional two clinical trials have recently tested the safety and immunogenicity of HIVA DNA-MVA vaccination in 48 healthy Gambian infants born to HIV-1-uninfected mothers (NCT00982579 trial) and in 72 healthy Kenyan infants born to HIV-1-infected mothers (NCT00981695 trial). These results are currently pending.
IMPROVEMENTS IN MULTI-EPITOPE VACCINES
Priming with a DNA vaccine and boosting with a viral vector vaccine has previously been shown to be an effective way of inducing immunogenicity. Nevertheless, despite the rational design and the use of a string of CTL epitopes, the HIVA prime-boost vaccination failed to induce strong CTL responses in the IAVI trials (Table 4). The string of CTL epitopes optimized for processing in a DNA vaccine (HVTN-048) as well as a peptide vaccine supplemented with adjuvant and GM-CSF (HVTN-056) also failed to induce the desired responses in the vaccinees. These results lead to concerns about the multi-epitope approach for designing an effective HIV vaccine. However, a number of methods for improving vaccine immunogenicity and stimulating CD8⁺ T cells have yet to be investigated in clinical trials. They include, and are not limited to the following: the enhancement of epitope by increasing the affinity or cross-reactivity of the epitopes for MHC and/or TCR [144]; the use of multiple cytokine combinations (GM-CSF + IL-12; CD86 + GM-CSF + IL-12; IL-15) [145-147]; the use of toll-like receptor (TLR) modulators such as CpG for DNA vaccines [148]; the use of alternate vaccine types and routes of administration [35,149-151] including autologous dendritic cells as a vaccine delivery system [144]. Several approaches might be used in combination, leading to improved immunogenicity of multi-epitope vaccine candidates.
Multi-epitope vaccines can be designed to include HLA supertypes for populations with a diverse genetic background, and promiscuous epitopes can help limit the number of final epitopes without decreasing the genetic coverage. Multi-epitope vaccines can include both dominant and subdominant epitopes, while avoiding antigenic elements that may not be favorable for protection. Even moderate successes with this type of vaccine against HIV might lead to improvements in future multi-epitope vaccine designs, not only against HIV but also against other pathogens.
SELECTION OF HIV IMMUNOGENS AND CONSERVED EPITOPES
Careful design of vaccine immunogens for protection against a wide number of HIV variants will be required to deal with the large antigenic diversity. Conserved viral antigens, subtype-matched antigens, consensus antigens, variants of single antigens and multiple antigens have all been used alone or in combination [152,153]. Table 4 shows a few examples for each of the strategies. Cross-clade CTL responses have been shown to preferentially target the conserved regions over the more variable ones [154], and these responses have been associated with better HIV disease outcomes or no disease manifestation [107,155,156]. The most conserved regions of HIV, especially those conserved across subtypes [153] or among lentiviruses [21], may be the best targets of the immune system for inducing vaccine protection. Some of these regions may be protective and are less likely to mutate because they hold a functional or structural importance to the virus species (possibly to the genus); a mutation would induce impairment to viral fitness [157,158]. This possibility makes the identification of conserved epitopes an important aspect of immunogen selection in vaccine design. One means of including these conserved regions is to construct polyvalent mosaic proteins as vaccine immunogens; thus far, preclinical evaluations of the mosaic vaccine have demonstrated great potential for broad T-cell responses, across subtypes [153,159,160].
A method of selecting highly conserved regions is to identify those with the lowest entropy, which is the lowest variability at each aa position. Based on this concept, the most conserved HIV proteins have been shown to be (in order of lowest variability): integrase (IN), core capsid (Gag-p24), reverse transcriptase (RT), and protease (PR) (Table 5) [161]. They were followed by Vpr, Vif, matrix (Gag-p17), Nef, Rev, and the surface envelope (SU-Env). Tat and Vpu have the highest variability (Table 5). This observation suggests that the selection of conserved vaccine epitopes should be done first from IN, Gag-p24, RT, and PR.
While Jenner may not have considered functional conservation when developing his smallpox vaccine, he can be considered to have been the first developer of a vaccine that was based on conserved features between two different viral species [162]. In a similar fashion, comparisons with other lentiviruses could help identify highly conserved epitopes that are required for viral function and survival. FIV is a lentivirus that is only distantly related to HIV-1, but may still be relevant to the evolutionary conserved approach of vaccine development because of the shared similarities between the HIV and FIV viruses in terms of aa sequence, structure, and pathogenesis [20]. A comparison of the aa composition of proteins between HIV-1 and FIV demonstrates the following percentages of identity/ homology: RT, 47/72; IN, 37/65; Gag-p24, 32/63; nucleocapsid (Gag-p7), 30/54; PR, 24/48; Gag-p17, 20/50; SU-Env, 19/43; transmembrane envelope (TM-Env) 18/42 [21] (Table 5). The three most conserved proteins are also those that have the lowest entropy calculation, as shown in Table 5 [161]. Hence, the IN, RT, and Gag-p24 proteins appear to be excellent targets for identifying evolutionary conserved regions that may also contain conserved T-cell epitopes.
IDENTIFICATION OF EVOLUTIONARY CONSERVED HIV CTL EPITOPES: USE OF FIV PROTEINS
Immunoninformatics has become an integral part in the design of new vaccines with great promise of rapid and effective vaccine discovery [163-165]. A number of tools and databases are now available online including HLA class-I and -II binding predictions [166,167], and a number of tools that are useful for the prediction of CTL epitopes (Table 6). In one study performed by our group, NetCTL-1.2 was used to identify CTL epitopes on the integrase sequences of HIV, SIV and FIV (Fig. 1A). For the twelve HLA supertypes shown in Fig. (1), a large number of CTL epitopes were predicted on each integrase sequence regardless of the virus: HIV with 78 epitopes, SIV with 74 epitopes, and FIV with 85 epitopes. Some of these were conserved between HIV and SIV (34 epitopes), as well as between HIV and FIV (25 epitopes) (Fig. 1B). A smaller number (17 epitopes) was conserved among all three viruses, reducing the target epitopes to the expected most evolutionary conserved.
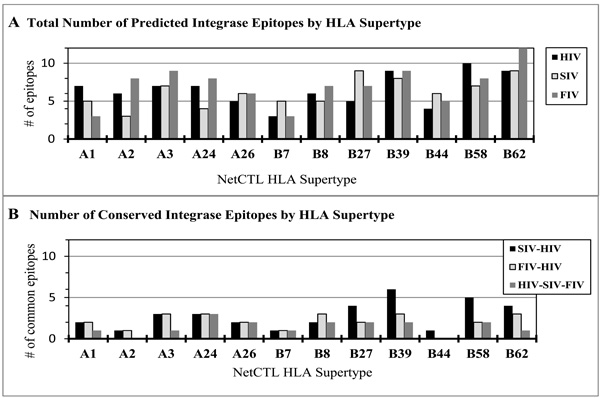
NetCTL-1.2 prediction of HIV, SIV, and FIV CTL epitopes. NetCTL-1.2, which is based on proteosomic C-terminal cleavage, TAP transport efficiency, and epitope binding to MHC class I alleles, was used to predict CTL epitopes shown by HLA supertypes (http://www.cbs.dtu.dk/services/NetCTL/).The total number of predicted epitopes by HLA supertype (A): HIV (78), SIV (74), and FIV (85) were tallied after analysis of the full-length integrase sequence from each virus. The predicted CTL epitopes were compared and the conserved epitopes between the viruses were identified based on aa position and same predicted HLA supertype (B): HIV-SIV (34), HIVFIV (25), and HIV-SIV-FIV (17).

Possible Location of Counterpart Epitopes.
HIV proteins (A, B) aligned to FIV proteins (A, B) showing four HIV epitopes (h1, h2, h3, h4) and three FIV epitopes (f1, f2, f3) with arrows indicating the location of the direct counterpart (arrow a) and indirect counterpart epitopes (arrows b, and c).
Thirteen HIV CTL epitopes termed best-defined CTL epitopes have been identified empirically on HIV integrase by different laboratories and compiled on the Los Alamos National Laboratory (LANL) website (Table 7). In this regard, based on observations using SIV and FIV, an evolutionary conserved HIV CTL epitope can be defined as a CTL epitope with a direct or indirect SIV and/or FIV CTL counterpart (Fig. 2). Using the direct counterpart approach (Fig. 2, arrow a), three of these epitopes are predicted to be CTL epitopes conserved between HIV, SIV, and FIV and one was shown to be an indirect FIV counterpart (Table 8). They share the same HLA binding and CTL supertype predictions [169,171]. As illustrated in Fig. (2), an evolutionary conserved epitope may be located on an HIV protein different from that of FIV (Fig. 2, arrow c). An indirect counterpart to an HIV epitope (bolded in Table 8), located upstream on FIV integrase, has higher aa identity and homology than the direct counterpart (Fig. 2, arrow b). This indirect FIV counterpart has the same binding alleles and predicted CTL supertype as the HIV epitope.
The predicted results of SIV sequences can be explained by the high aa identity between HIV and SIV as SIV is more closely related to HIV than FIV. However, despite the relatively lower aa identity between HIV and FIV, FIV counterpart epitopes still appear to be potentially effective HIV antigens (see Table 8), most likely due to the slightly higher aa homology observed between the two viruses. This finding indicates the strong potential that both SIV and FIV epitopes could induce CTL responses in human PBMCs and suggest that this type of comparison may lead to the identification of evolutionarily conserved CTL epitopes on HIV. Therefore, conserved SIV and FIV IN peptides can be used as immunogens in vitro to compare and identify conserved immune responses generated by the PBMCs of HIV⁺ individuals.
CONCLUSION
An effective prophylactic HIV vaccine should probably include both humoral and CMI responses, since antibodies (bNabs, ADCC antibodies) and CTLs are likely to play critical roles in the control of the virus. HIV epitopes that are highly conserved are believed to be relevant to viral fitness and thus important in the design of a global HIV vaccine. The identification of evolutionary conserved linear epitopes is feasible for both B and T cells with currently available bioinformatic tools, but most bNAbs do not usually target linear epitopes. Therefore this approach is more useful for identifying T-cell epitopes. Testing for immune responses in HIV⁺ individuals and vaccinated volunteers from clinical trials can help determine the relevance of these epitopes in protection against the virus. Conserved CTL epitopes could be used to supplement HIV protein vaccines or used as part of a prime-boost combination, which has so far provided the best efficacy in a phase-III trial. An effective prophylactic T-cell vaccine containing highly conserved epitopes may inform therapeutic strategies and help the millions of HIV-infected individuals to effectively control viral replication with or without ART.
CONFLICT OF INTEREST
J.K.Y. is the inventor of record on a patent held by the University of Florida and may be entitled to royalties from companies developing commercial products related to the research described in this paper. Dr. Yamamoto is the inventor of record on a University of Florida patent and may be entitled to royalties from companies developing commercial products related to the research described in this paper.
ACKNOWLEDGEMENTS
This work was supported by NIH R01-AI65276, R01-AI30904, and JKY Miscellaneous Donors Fund.

