All published articles of this journal are available on ScienceDirect.

Evolutive Temporal Footprint of an HIV-1 Envelope Protein in an Epidemiologically Linked Cluster
Authors Info & Affiliations
Abstract
Background:
The C2V3C3 region of gp 120, encoded by the HIV-1 envelope gene (env), is an important antigenic target, a key determinant for viral evolution and essential for determining epitopes for vaccines.
Methods:
The relationships among genetic sequence diversity, selective pressure, constraints on HIV-1 envelope protein were explored and also correlated this analysis with information entropy; hypermutation; HIV tropism; CD4+ T cell counts or HIV viral load. A total of 179 HIV-1 C2V3C3 sequences derived from cell-free plasma were used, determined from serial samples, in four epidemiologically linked individuals (one infected blood donor, two transfusion recipients and a sexual partner infected by one of the recipients) over a maximum period of 8 years. This study is important because it considers the analysis of patterns in genomic sequences, without drugs and over time.
Results:
A temporal relationship among information entropy, hypermutation, tropism switch, viral load, and CD4+ T cell count was determined. Changes in information entropy were time-dependent, and an increase in entropy was observed in the C2V3C3 region at amino acids G313 and F317-I320 (related to the GPGR-motif and coreceptor tropism), and at amino acids A281 in C2 and A346 in C3, related to immune escape.
Conclusion:
The increase of information entropy over time was correlated with hypermutation and the emergence of nonR5- strains, which are both associated with more variable genomes.
HIGHLIGHTS
√ Increased information entropy correlates with a tropism switch from R5 to non-R5 strains.
√ Increased information entropy correlates with decreased CD4+T cell count and viral load, and increased hypermutation.
√ HIV genetic evolution and the selection of escape mutants result in the emergence of more disorganized HIV genomes.
1. INTRODUCTION
The ability of HIV-1 to rapidly evolve is one of its most striking features, enabling its immune escape and favoring its persistence in an infected host [1, 2]. The HIV-1 envelope gene (env) encodes the viral envelope glycoprotein, which is a heavily glycosylated trimer of non-covalently linked, heterodimeric glycoproteins composed of the surface-exposed gp120 and the transmembrane gp41. Extensive N-linked glycosylation of the envelope glycoprotein effectively shields many of its conserved epitopes from antibody recognition. Based on sequence variability, gp120 is divided into five conserved regions (C1-C5) and five hypervariable loops (V1-V5). Hypervariable loop 3 (V3) of gp120 is approximately 35 amino acids long, frequently glycosylated, and highly variable and has a disulfide-bonded structure that has potential impact on several functions of the envelope protein. The V3 loop harbors the most important determinants of viral tropism (i.e., coreceptor usage), as well as major antigenic neutralizing epitopes [3-6].
The relevance and novelty of this study lie in the analysis of patterns in genomic sequences, by using information metrics as well as the variability accumulated occurrence, in the natural history of HIV infection over time, in an epidemiologically linked cluster once a few studies address this kind of approach. Thus, the analysis using informational metrics lies in quantifying the genomic patterns over time, which is fundamental to study and planning of new targets based on epitopes behavior. In this sense, the present study analyzed the intrapatient evolution of the gp120 C2V3C3 region in four distinct epidemiologically linked individuals over the course of approximately 7 years, focusing on evolutionary selective pressure and its association with information entropy, hypermutation, tropism and clinical markers of disease progression [7-11]. When used in genomic analysis, information entropy brings important insights into the evolutionary relationship between the virus and its host, which is essential for designing new antiretroviral drugs and vaccine candidates against HIV, and also provides insights into virus persistence. Since genomic evolution is a process that enables escape from environmental constraints imposed by the immune system or antiretroviral [12-18], we evaluated information entropy over time and its association with HIV-1 evolution as inferred from genetic diversity.
2. METHODS
2.1. Patients and Sequences
The sequences used in this study were obtained from a cluster of four HIV-1-infected individuals comprising a blood donor, two recipients of infected blood from the donor, and a sexual partner infected by one of the recipients [19]. The patients enrolled in this study were at an asymptomatic stage of infection and were antiretroviral-naïve adults. Sequence data were obtained from serum from the blood donation sample and from plasma samples collected from the four members of the epidemiologic cluster over time. Sequences derived from cell-free plasma were determined from serially collected samples. Briefly, viral RNA from plasma samples collected from the four members of the epidemiological cluster over time was used to generate molecular clones by single genome PCR amplification from viruses isolated at distinct time-points, and the sequences were generated from a section of env that includes the 3’end of the C2-encoding region, the V3-encoding region, and the 5’end of the region that encodes C3. Nine or more sequences per patient per time-point were obtained, which yielded a total of 179 sequences. The number of samples was not constant; a range of 7-11 clones per segment were sequenced to obtain a total of approximately 49 sequences per patient. The C2V3C3 env region HIV-1 subtype B sequence data were from samples collected over the 8-year period from 1986 to 1993, and the analyzed env fragment encodes 76 residues corresponding to amino acids F277 to F353 of the HXB2 reference strain. The sequences are from an epidemiologic cluster that is composed of a blood donor (D.O.) who donated HIV-infected blood in 1985, two recipients (R.A. and R.B.) infected by the contaminated blood, and one individual who was infected by sexual contact (S.C.) with R.B. These individuals remained antiretroviral naïve during the entire follow up of seven years; thus, the molecular evolution depicts natural progression in the host. The individuals remained asymptomatic during the course of the study and had stable and normal CD4+ and CD8+ counts and low viral loads. HIV-1 plasma viremia was quantified with a quantitative competitive RT-PCR assay as we can see on [19]. All sequences reported in this publication were submitted to GenBank (accession numbers U29433–U29437, U29956, U29957, U29959–U30074, U30077–U30145, U31573–U31582, and U43035–U43054). For further analysis, the sequences of each patient were grouped according to collection year and aligned using ClustalW [20] with sequence D85.40 (from D.O.) as a reference because it represents the strains exclusively found in the blood inoculum. Sequencing errors, genomic regions with deletions or insertions were excluded from the analysis to preserve the reading frame.
2.2. Evolutionary Analysis
We analyzed the evolution pattern of the C2V3C3 regions of HIV-1 gp120 over time by determining information entropy and its relationship with tropism, hypermutation, viral load, and CD4+ T cell count. Entropy is a standard measure to evaluate protein variability that quantifies the uncertainty of information per site or position in the genome and considers the number of possible amino acid replacements and their frequency. Information entropy (H) is commonly used to quantify the uncertainty of information about the amino acid or protein at a given position and their fixation over time. The classical Shannon formula for the entropy, or information content, per position of the amino acid sequence, is written as:
Equation 1:Information Entropy [21].
 |
(1) |
where p(x) is the probability of the base (A, T, C, G) in the given sequence; in other words, p(A) is the probability of the occurrence of the base “A”. The probability was estimated by the frequency (by counting the occurrence) of each amino acid (a.a.), x represents each of the possible a.a. and log2 represents the logarithm in bits. A value of 0 for H indicates that all sequences are identical at a given position, whereas a non-zero value indicates that different amino acids are present. The average information entropy over time at each position in the C2V3C3 regions of HIV-1 gp120 was determined using MATLAB software [22]. An information entropy value less than 0.2 was considered conserved, a value between 0.2-0.5 was considered semi conserved, and a value above 0.5 was considered nonconserved [23, 24]. Hypermutation/G→A substitution analysis of the HIV sequences was performed using the Hypermut 2.0 tool, available at LANL HIV Sequence Database [25], and the coreceptor tropism of the sequences was evaluated by the Geno2pheno algorithm, available at http://coreceptor.geno2pheno.org/index.php [26], with a False Positive Rate (FPR) cutoff of 10% in accordance with European guidelines [27].
2.3. Analysis and Data Validation
To quantify the relationships among the evolutive patterns of the envelope genes of the group, Pearson’s correlation coefficient was calculated for the time series data. The relationship among information entropy, CD4+T cell count, viral load, tropism, and hypermutation in the group was calculated by pairwise correlations matching each genome position, and the corresponding P value of each measurement, hypothesis test, entropy means, and R2 were determined. Statistical analysis was performed using Excel®, MATLAB and GraphPad Prism version 6.0 software [22, 28]. We considered a P value of 0.05 to be statistically significant, aiming to a more conservative analysis.
3. RESULTS
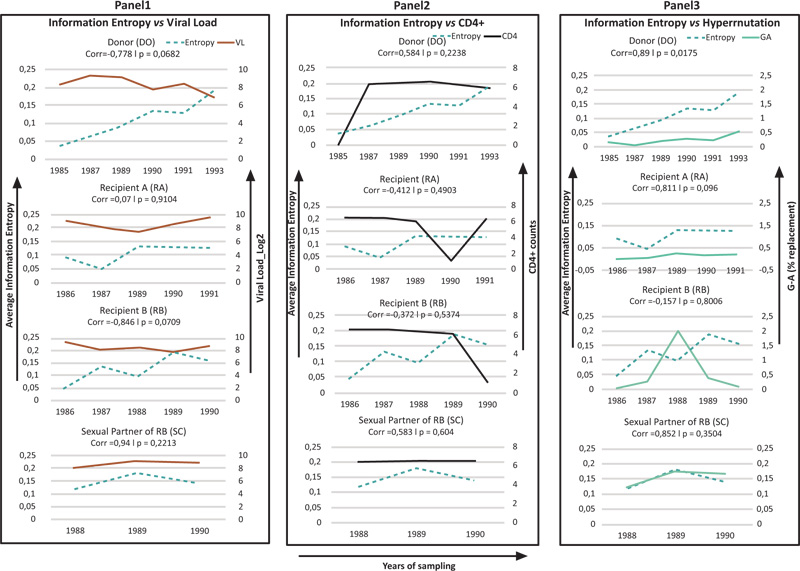
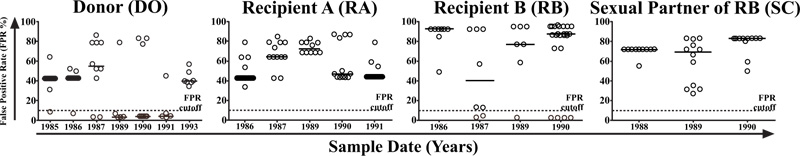
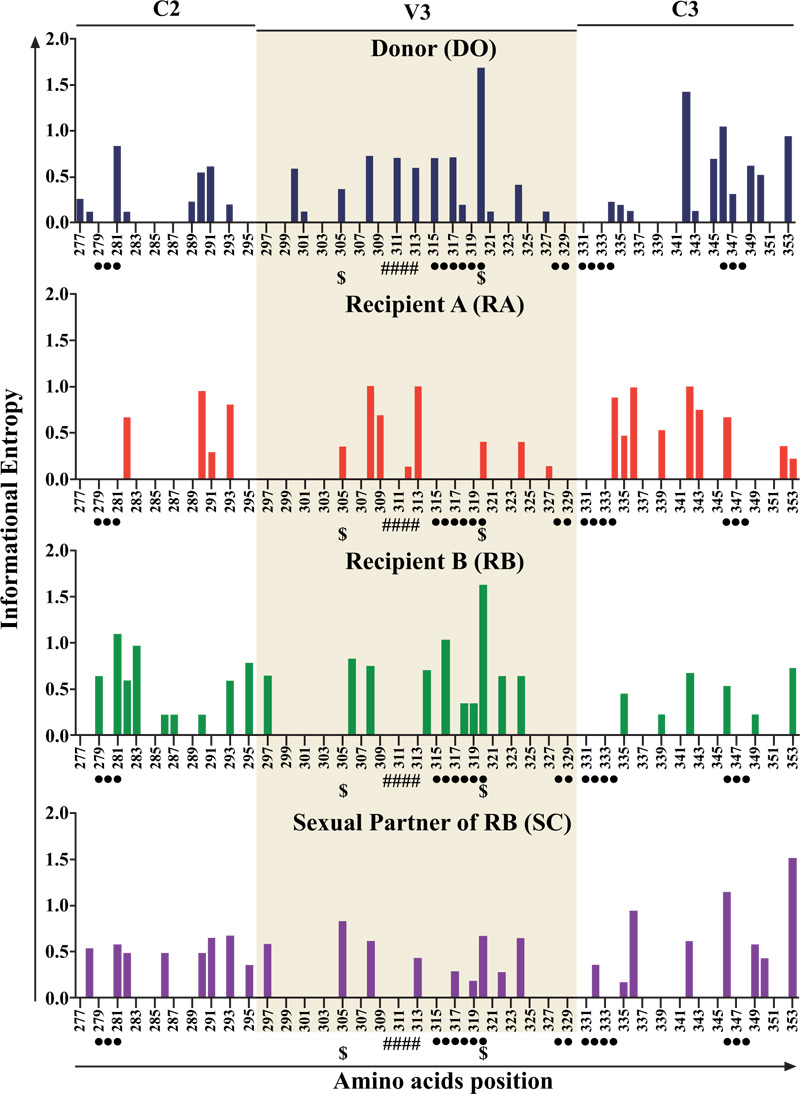
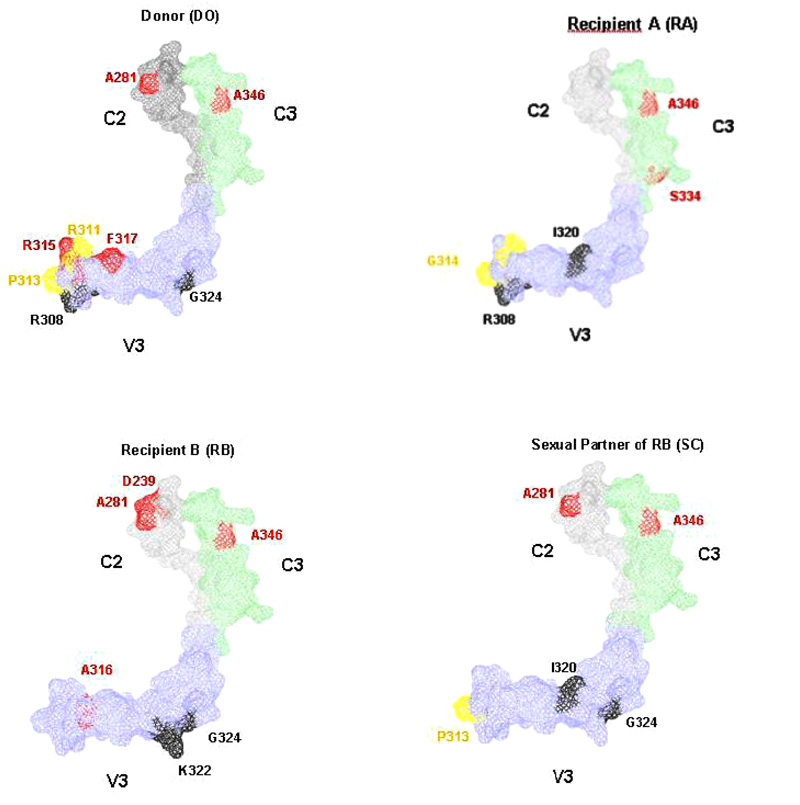
Information entropy showed a progressive increase in all individuals in this cohort over time. A correlation between information entropy and viral load was observed in all individuals but was statistically significant only in D.O.(p<0.007) (Fig. 1, Panel 1). Similarly, information entropy correlated with CD4+T cell count in patients D.O. (p=0,223), R.A. (p-0,490), R.B. (p=0.537), and S.C. (p=0,604). Additionally, R.B. presented a decrease in CD4+T cell count associated with an increase in entropy (Fig. 1, Panel 2). To obtain further insight into molecular evolution, we analyzed the relationship between information entropy and hypermutation. In an analysis of G-A hypermutation vs entropy, a significantly positive correlation was seen in D.O.(p=0.017) and R.A. (p=0.096), but no statistically significant correlation was seen in R.B.(p=0.800) or S.C.(p=0.350), as in Fig. 1, Panel 3. Only D.O. and R.B. harbored a small proportion of CXCR4-tropic strains (i.e., non-R5), which were present from the beginning of the infection (Fig. 2). We, therefore, inspected the relationship between information entropy and G-A hypermutation and the presence of non-R5 strains. The progressive increases in entropy and hypermutation in D.O. and R.B. could have resulted in the emergence of non-R5 strains and, in the case of R.B., disease progression. R.A. did not present any AIDS-defining opportunistic infections or neoplasms during the follow-up period, although the CD4+T cell decay clearly demonstrated HIV-1 disease progression, whereas R.B. died in 1991 because of AIDS. Additionally, non-R5 strains were identified in D.O. in 1993 and in R.B. as early as the second evaluated time point in 1987 (Fig. 2). Here, we utilized the Geno2pheno[coreceptor] algorithm to determine coreceptor usage among the analyzed genomic sequences. We observed an increase in information entropy over time and emergence or increase in the prevalence of non-R5 strains over time when single genome amplification methodology was used. In addition, when analyzing the average information entropy at each amino acid position in C2V3C3 of gp120 computed over time, we observed a persistent increase in information entropy in all individuals (Figs. 3 and 4). Genomic variations differed substantially among individuals at later time points. Table 1 summarizes the amino acid positions that remained conserved (entropy score <0.20) or were associated with viral evolution (entropy score >0.5) over time. D.O. showed higher entropy in the V3 region at a.a. K305, R311, P313, R315, F317 and I320 (related to tropism, immune escape and GPGR-motif), in the C3 region at a.a. A346 (related to immune escape) and in the C2 region at a.a. A281 (related to immune escape) (Fig. 3). R.A. showed more entropy in the C3 region at a.a. G313 and S334-A336 (related to GPGR-motif and immune escape) [29, 30]. R.B. showed higher entropy in the V3 region at a.a. A316 and I320 (immune escape and tropism, respectively), as well as in the C2 region at a.a. D279, A281 (immune escape) and in the C3 region at a.a. A346. S.C. showed higher entropy in the C3 fragment at a.a. A346 (immune escape), in the V3 region at a.a. K305, P313 and I320 (related to tropism and GPGR-motif), and in the C2 region at a.a. A281 (related to immune escape) [31]. Similarly, several residues were identified as common in all individuals of the cohort and remained conserved (information entropy equal to zero or <0.2): a.a. N280, T283-I285, and L288 in C2,a.a. R298, R304, I309, and Q328-I333 in V3, and a.a. E351 and Q352 in C3 [29-32].
| Amino Acids | ||
| ID | Information Entropy <0.20* | Information Entropy >0.50* |
| D.O. | D279, N280, T283-L288, V292, I294, P299, N302-R304, R306, I307, I309, Q310, Q328-I333, K337-T341, E351, Q352. | A281, T290, S291, N300, K305, R308, R311, P313, R315, F317, I320, G324, L342, I345, A346, L349, R350, F353. |
| R.A. | F277-A281, T283-289, I294-R304, G314-T319, G321-I323, Q328-I333, S347-E351. | K282, T290, S291, E293, K305, R308, I309, G313, I320, G324, S334-A336, N339, L342, K343, A346. |
| R.B. | F277, T278, I284, I285, L288, N289, R298-K305, I309-P313, R315-S334, K343-I345, R350-Q352. | D279, A281, T283, E293, N295, T297, R306, R308, G314, A316, I320, K322, G324, R335, L342, A346, F353. |
| S.C. | D279, N280, T283-I285, Q287-N289, R298-R304, I309-G312, G314-A316, N325-C331, K337-T341, K343-I345, S347, E351, Q352. | T278, A281, K282, V286, T290, S291, E293, N295, T297, K305, R308, P313, I320, G324, A336, L342, A346, L349, R350, F353. |
4. DISCUSSION
This study describes the relationship between informational entropy in C2V3C3 of Gp120 and correlates it with APOBEC-driven G-A substitutions, viral load, CD4+T cell and predicted tropism. The informational entropy correlation with CD4+T cell count (Fig. 1, Panel 2) similar to another study, that demonstrated that less organized HIV genomes, as inferred from higher levels of information entropy, correlate with less competent host immune systems [24]. Non-R5 strains have been associated with the presence of positively charged amino acids in the V3 loop of gp120. It has also been described that, in contrast to CCR5, the CXCR4 coreceptor is filled with negatively charged amino acids, which better enables the attachment of dual-tropic HIV strains [33, 34].
Interestingly, positively charged amino acids, such as arginine and lysine, are usually coded by nucleotide triplets such as AGA and AGG (arginine) or AAA and AAG (lysine), which have large amounts of As. Non-R5 HIV-1 strains may emerge early in the HIV infection and be associated with faster HIV-1 disease progression [35-37]. We utilized the Geno2pheno [coreceptor] algorithm to determine coreceptor usage among the analyzed genomic sequences. Although phenotypic assays are the gold standard for determining coreceptor tropism, genotypic assays provide a reasonable alternative and have been increasingly used. Additionally, the European Consensus Group considers genotypic assays sufficient prior to prescribing maraviroc. Among the different coreceptor tropism prediction algorithms available online, Geno2pheno [coreceptor] has demonstrated comparable performance to the original and enhanced-sensitivity Trofile phenotypic assays [26, 27, 38-40].
Additional evidence for the validity of Geno2pheno [coreceptor] predictions is based on the fact that, in an individual patient, the FPR values tend to progressively decay, leading to the emergence of CXCR4-tropic strains with a mean evolution time of 27.29 months (range, 8.90 to 64.62) when populational sequencing is used. In addition, increases in entropy over time observed here are indicative of viral evolution with discrete escape and stabilization of the viral population. Amino acids with high entropy scores are located within the core of the envelope protein. Gp120 variations are not uniform; occur at different frequencies and in different fragments of HIV-1 gp120, and allegedly influence protein folding. Conserved residues, as seen in C2V3C3, are responsible for viral structural maintenance and replication (Fig. 3). Interestingly, in all individuals of this cohort, amino acids A281, 282, 290-293, 295, 305, 308, 313, 320, 324, 342, 343, 346, 350, and 353 (related to immune escape, T cell recognition epitopes, and tropism, were constantly under environmental disturbance or in an adaptive process [29-32, 40-43]. Additionally, with the exception of the result on R.B. (Fig. 1), the increase in entropy is proportional to increases in hypermutation and tropism change; it is also associated with the structural genomic order, and consequently, the structural and/or functional stability of DNA.
Although we have performed analysis using molecular clones in a temporal evolutive fashion, we recognize that this work has a serious limitation in scope since the data set is derived from only 4 HIV infected patients. Nonetheless, based on the obtained results, we can infer that these conserved residues are of interest for future studies aimed at the design of epitopes and the development of therapeutic molecules to treat HIV. In this context, these results highlight the constraints on the evolution of the same viral quasispecies present in distinct human hosts over time.
CONCLUSION
We identified important functional constraints related to evolution in the genomic regions C2V3C3 of HIV-1 gp120. The genomic variability occurred in specific residues related to immune escape was time-dependent among all individuals and showed a direct relationship with viral evolution, escape fitness and disease progression (D279, A281, R315, A316, F317, S334, A346; R308, I320, K322, G324; R311, P313). Moreover, according to the results seen in one of the patients and considering the literature [36], we can speculate that HIV evolution, leading to the emergence of less organized genomes, will lead to the emergence of hypermutated HIV strains and, consequently, more cytopathic non-R5 strains develop, potentially contributing to a rapid disease progression for HIV.
Thus, understanding the evolutionary pattern of HIV-1 is critical, and genomic analysis using information entropy provides important insights into evolution and genetic restriction and the conserved regions identified in this study that can inspire designers of new molecules and vaccines based on conservation patterns.
AUTHORS’ CONTRIBUTIONS
Designed the research: E.N.d.C.L., R.S.D., and J.R.C.P. Performed and interpreted the experiments in evolutionary analysis and information entropy: E.N.d.C.L., R.S.A.L., and J.R.C.P. Performed and wrote about tropism analysis: M.A.S. Wrote the final manuscript: E.N.d.C.L., R.S.D., and J.R.C.P. All authors read and approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The Federal University of Sao Paulo, Brazil, Ethics Committee and Human Research Ethics under number #0024/11 approved this study in February 2011, and the Brazilian Ministry of Health authorized it.
HUMAN AND ANIMAL RIGHTS
No animals were used in this research. All human research procedures followed were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Informed consent has not been obtained for this study since HIV genomic sequences were generated for clinical purposes.
AVAILABILITY OF DATA & MATERIALS
The authors confirm that the data supporting the findings of this study are available within the article.
FUNDING
This work was financially supported by Fundação de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP), research grant # 2011/12156-0 to R. S. D. and a PhD scholarship from Coordenação de Aperfeicoamento de Pessoal de Nıvel Superior (CAPES) to E. N. d. C. L. The funders had no role in study design, data collection or analysis, the decision to publish or the preparation of the manuscript.
CONFLICT OF INTEREST
The author declares no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

