All published articles of this journal are available on ScienceDirect.

Further Evidence that Human Endogenous Retrovirus K102 is a Replication Competent Foamy Virus that may Antagonize HIV-1 Replication
Authors Info & Affiliations
Abstract
Objective:
The goals of the research were to determine if a foamy effect on macrophages was due to human endogenous retrovirus K102 (HERV-K102) replication, and to further address its potential significance in HIV-1 infection.
Methods:
An RT-PCR HERV-K HML-2 pol method was used to screen the unknown HERV, and isolated bands were sent for sequencing. Confirmation of RNA expression was performed by a real time quantitative PCR (qPCR) pol ddCt method. Rabbit antibodies to Env peptides were used to assess expression by immunohistology and processing of Env by western blots. A qPCR pol ddCt method to ascertain genomic copy number was performed on genomic DNA isolated from plasma comparing HIV-1 exposed seronegative (HESN) commercial sex workers (CSW) to normal controls and contrasted with HIV-1 patients.
Results:
HERV-K102 expression, particle production and replication were associated with foamy macrophage generation in the cultures of cord blood mononuclear cells under permissive conditions. A five-fold increased HERV-K102 pol genomic copy number was found in the HESN cohort over normal which was not found in HIV-1 positive patients (p=0.0005).
Conclusions:
This work extends the evidence that HERV-K102 has foamy virus attributes, is replication competent, and is capable of high replication rate in vivo and in vitro. This may be the first characterization of a replication-competent, foamy-like virus of humans. High particle production inferred by increased integration in the HESN cohort over HIV-1 patients raises the issue of the clinical importance of HERV-K102 particle production as an early protective innate immune response against HIV-1 replication.
INTRODUCTION
The human endogenous retrovirus-K (HERV-K) HML-2 group is the most recently integrated and most biologically active of the endogenous retroviruses [1]. The group consists of two types: those with a 292 bp nucleotide sequence encoding a Rec-like domain are type 2; while those lacking this domain are type 1 and encode np9 instead [1]. It has been suggested that type 1 HML-2 proviruses would not be able to replicate due to the lack of the Rec-like domain [1]. As well, it has been purported a naturally-occurring, replication competent HERV has not been described [1,2], and that no HML-2 particles are produced in HIV-1 patients [2]. However, our previous published data is inconsistent with the validity of these concepts [3].
We previously published that particles from HERV-K102, a type 1 HML-2 provirus, contain predominantly DNA genomes as well as morphological evidence consistent with HERV-K102 being replication competent by virtue of its foamy virus-like properties [3]. Foamy viruses (FV) are unconventional retroviruses with a reverse life-cycle to orthoretroviruses, and despite lacking a Rec-like domain, are replication competent and fully infectious [4-8]. In this previous study, HIV-1 patients had lower maximal HERV-K102 replication levels than patients harboring other bloodborne pathogens [3]. However, from the data provided in table 3 of ref. [3], HIV-1 patients produced on average about 8,200 HERV-K102 DNA-bearing particles per ml of plasma and where 72% of this subgroup of HIV-1 patients had demonstrable particles. Importantly, in the HIV-1 pati-ents, the excess DNA signals found in plasma were conf-irmed to be cDNA and not genomic material. In contrast, HML-2 or HERV-K102 virions were not found in 30 of 30 normal adults. Thus, it appeared that HIV-1 and other bloodborne pathogens induce HERV-K102 particle production in vivo [3]. In addition, HML-2 particles were also isolated in adults with active disease but not when they were in remission. Finally, virions were also detected in 50% of normal cord blood plasma samples. All together these results suggested that HERV-K102 particle production may relate to innate immune activation, and the replication strategy of HERV-K102 might be more analogous to foamy retroviruses than orthoretroviruses, explaining how it could replicate without a Rec-like domain [4-8].
Others have since characterized type 1 and type 2 HERV-K HML-2 particles in the plasma of HIV-1 patients [9,10]. It was also shown HIV-1 Tat may also have induced HML-2 expression in vitro [11] which supports our in vivo findings reported earlier [3]. More recently, Markovitz’s research team demonstrated reconstituted, DNA-bearing HERV-K HML-2 particles were replication competent and infectious in vitro [12] in direct substantiation of our in vivo finding that replication competent HERV-K102 had cDNA genomes [3]. Therefore it seemed the conclusions reached by Coffin and colleagues [1,2] that no HML-2 virus replicates in HIV- patients, may have been largely unfounded.
Indeed, with respect to the unexpected conclusions in the paper by Bhardwaj et al. [2], while the data in HIV-1 patients were convincing for the lack of HML-2 particles with RNA genomes, they reported the HML-2 DNA signal as determined on isolated particles was statistically higher in HIV-1 patients than in controls. Surprisingly, the GAPDH controls for genomic DNA were performed but were not utilized to gauge background levels of genomic DNA in the isolated particles. At face value, and assuming the GAPDH values were the same in both HIV-1 and controls, one could calculate from their published data [2], that on average, HIV-1 patients had about 8,300 DNA-bearing HML-2 particles per ml of plasma and 73% produced particles, in direct validation of our findings. Indeed the close agreement of our findings and our interpretation of the Bhardwaj et al. results, suggests that HERV-K102 DNA-bearing particles might be the only HML-2 particles produced in HIV-1 patients. Nonetheless, the existence of replication competent DNA-bearing HML-2 particles in HIV-1 patients is not widely appreciated and the significance of the relatively low levels of HERV-K102 particles in chronically infected HIV-1 individuals remained unclear.
Our hypothesis is that HERV-K102 is a foamy-like virus. Aside from being non-pathogenic, FV have several other features distinguishing them from orthoretroviruses. They can integrate to high proviral copy number such as up to 20 proviral copies per genome [13]. Env expression is absolutely required for particle budding into vacuoles, and processing of Env into the surface unit (SU) and transmembrane domains by cellular furins are essential for infectivity [14]. Heterologous Env cannot substitute in the particle during morphogenesis [14]. Paradoxically, they are non-pathogenic yet lyse certain cell lines but not others in vitro [5-7]. FV have been identified in many species except humans [6]. It should be noted that the previously characterized human foamy virus was discovered to have originated in chimpanzees and was renamed prototypic foamy virus (PFV) [6]. The role of FV in health or human disease remains largely unknown.
During the set-up of human cord blood mononuclear cells (CB) as indicator cells for studying potential xenozoonotic agents, it was serendipitously discovered by electron microscopy that in control cultures high levels of immature retroviral particles were budding into vacuoles which gave the cultured CB a foamy appearance when cells were cultured in IMDM, but not when cultured in RPMI. The goals of this research were to determine the relationship of the foamy CB and replication of HERV-K102, and to attempt to address the clinical significance of HERV-K102 particles in HIV-1 infected patients.
METHODS
Sample Collection and Cell Culture
Ethics approval, anonymous sample collection, culture and storage were previously described for these studies [3]. In brief, cord blood samples were collected in heparinized tubes and cord blood mononuclear cells (CB) were isolated using Ficoll-Paque standard protocols (GE Healthcare Bioscience Inc., Baie d’Urfe, Quebec, Canada). CB were cultured at 5 x 105/ml in Iscove’s Modified Dulbecco’s media (IMDM) with 10% fetal calf serum (FCS) at 37oC for 7 days unless noted otherwise. The HIV-1 exposed seronegative (HESN) plasma samples representing commercial sex workers (CSW) who had shown at least 3 years of resistance to HIV-1 transmission despite repeated exposures were a kind gift from Drs. Francis A. Plummer and Keith Fowke and had been described previously [15,16].
ML4 and ML5 Rabbit Antibodies to HERV-K102 Env
The ML4 peptide has the amino acid sequence KRASTE MVTPVTWMDN (GenBank accession # AF164610) and is common with a number of type 1 HERV-K (HML-2) family members but not with type 2. The ML5 peptide might be specific to HERV-K102, and has the sequence LETRDCKPF YTIDLNSS. Peptides and rabbit antisera made to the peptides were manufactured by Washington Biotechnology Inc., were affinity purified and adjusted to 1 mg/ml as described earlier [3]. The control rabbit antibodies were pooled from the pre-serum of the rabbits used for immuni-zation and were purified by isolation on Protein A. The ML4 and ML5 peptide epitopes are antigenic in humans and are recognized by sera from about 75% of HIV-1 patients [3].
Electron Microscopy
Cultures were harvested, washed three times in PBS (no FCS), and then placed in 4% paraformaldehyde fixative, pH 7.4 for 1 to 2 hours. The samples then went to the EM processing laboratory at the Children’s Hospital of Eastern Ontario.
Cytospins
1 x 106 CB cells were resuspended in 100 μl of 5% FCS -PBS and then applied to the cytospin holders. The samples were centrifuged at 600 rpm for 10 minutes. Air dried cytospins were fixed for 10 seconds in absolute ethanol. The slides were then immersed in Harris Haematoxylin for 3 minutes. The slides were washed in water (until it ran clear), and decolorized in 0.5% acid alcohol for 40 seconds. After washing in water for 5 minutes, the slides were stained in 1% Eosin for one minute, washed in cold water for 1 minute, then dehydrated (dipped sequentially in 3 alcohol containers), cleared by dipping the slides in 3 changes of toluene, and mounted.
HML-2 pol PCR and RT-PCR
The TRI Reagent Protocol was followed according to manufacturer’s instructions (Sigma-Aldrich T9424) for the preparation of RNA and DNA from cultured CB. Our novel primer set for HML-2 pol and the complete method was detailed previously [3].
HERV-K102 pol Quantitative Real Time ddCt PCR Method
We used our novel primer and probe set as described earlier, which uses 18 S RNA (Applied Biosystems Inc., Catalog #4331182, Hs99999901_s1) that can be used to internally standardize either for RNA or DNA testing [3]. For RNA isolation from cultured cells, the QIAamp RNA Mini Kit was used with the DNase digestion step and RNA clean up steps according to manufacturer’s instructions. For the RNA, we used a one-step reverse transcriptase procedure (Applied Biosystems Inc., protocol) where our specific primers were used for the reverse transcriptase step. Parallel analysis of RNA was conducted in Amperase–UNG in the Master-Mix buffer, which digested the products made during the reverse transcriptase step, to control for potential contaminating genomic DNA. For the RNA analysis the stock reference material for the ddCt was RNA from Applied Biosystems Inc., (50 μg/μl). For DNA isolation as performed on plasma samples [3], the QIAamp DNA Mini Kit was used and followed manufacturer’s instructions. The Master-Mix buffer employed Amperase–UNG to digest any cDNA present in plasma such as from HERV-K102 particles as previously described [3]. The reference DNA used for the ddCt method was from Applied Biosystems Inc., (male DNA 10 ng/μl). All real time PCRs were performed in triplicate. Our methodology had a detection limit for HERV-K102 pol of 25 copies to 1011,a co-efficient of variance less than 0.1%,and the primers and probe sequences were provided previously [3].
Immunohistology
CB (1 x 107) cultured for 7 days (Day 7) or freshly isolated CB cells (Day 0) were washed three times in PBS, pelleted, then fixed in 4% paraformaldehyde pH 7.4 for 1 to 2 hours. The cells were centrifuged, and the fixative replaced with PBS. Standard paraffin embedding for cell block preparation used agar in PBS (0.1 M pH 7.4) as a pre-supporting material for the cells. The cells were processed as usual in an automatic tissue processor (ATP Tissue Processor from Triangle Biomedical Systems Inc.). Paraffin blocs were cut at 4 microns, deparaffinized, hydrated and stained. For staining the section were incubated for one hour with ML4 or ML5 affinity purified rabbit antibody (1 mg per ml) or control purified rabbit antibodies diluted 1/1000 and then the Vectastain ABC Kit Immuno-peroxidase (Rabbit IgG) (Catalog # PK-4001, Vector Laboratories) was used following manufacturer’s instructions.
Combination Immunoprecipitation and Western Blotting
CB cells cultured for 7 days (Day 7) or not cultured (Day 0) were harvested and 5 x 107 cells were extracted in 500 μl lysis buffer (1% NP40, 100 mM NaCl, 10 mM EDTA in 100 mM TRIS-HCl pH 7.6), for 30 to 60 minutes at room temperature. The lysate was vortexed and centrifuged for 10 minutes at 250 x g to remove nuclei. The supernatant was retained. The supernatant was clarified by centrifugation for 30 minutes at 10,000 x g (microfuge). The lysates were first pre-treated to remove non-specific binding by adding 10 μl of goat anti-rabbit Ig agarose (RDI Research, product # 941-9136) per 200 μl of lysate. After one hour of incubation at 4oC with shaking, the tubes were centrifuged briefly at 200 x g (5 seconds) in a microfuge, and supernatants harvested. To 200 μl of treated lysates, 800 μl of Tris- buffered saline (TBS) was added and then purified ML4, ML5 or control rabbit antibody was added (10 μl of 1 mg/ml stock), and was incubated overnight at 4oC. The next morning, 50 μl of 1 mg/ml solution of agarose conjugated with goat anti-rabbit antibody was added and the tubes mixed with gentle shaking for one hour at 4oC. The agarose was then washed three times with 1 ml TBS by pelleting in a microfuge at 200 x g for 5 seconds with a final wash in 0.5 M TRIS, pH 6.8. The agarose was again pelleted, all the supernatant carefully removed and discarded, and 50 μl of Sample Buffer was added (25% glycerol, 2% SDS, 0.01% Bromophenol Blue in 62.5 mM TRIS-HCl pH 6.87). The samples were mixed and then heated at 100oC for 5 minutes. The tubes were microfuged briefly (5 seconds at 200 x g) to pellet the agarose, and 1 μl of the supernatant was loaded onto 10-15% SDS-PAGE gradient gels (Amersham BioSciences, pre-cast gels, Cat. # 170-0516-01). The samples were run on the Phast-Gel System (Pharmacia) according to manufacturer’s instructions. In order to validate proper transfer and to determine the molecular weight of bands revealed in the western blotting step, Coomassie Blue, pre-stained molecular weight standards (Biorad, 7 to 207 kilodaltons, Cat. # 161-0318) were also loaded onto the gradient gels after boiling. As soon as the SDS-PAGE separation was complete, pre-wetted nitrocellulose (Biorad) was placed onto the gels, and the positions of the molecular weight standards were pencilled in. Care was taken to avoid transblotting the stacking gel. The transblot was assembled according to manufacturer’s instructions with filter papers pre-wetted with transblot buffer (21.25 g of Na2HPO4-7H2O and 5.52 g of NaH2PO4 added to 4 liters of ddH2O, pH 7.4), and the transblot console placed onto the separation bed of the Phast-gel system. The transblot was run for exactly 17 minutes. After completion of the transblot, the nitrocellulose was removed, washed in one change of dd H20, then TBS, and then incubated with primary antibody (affinity purified ML-4) diluted 1/1000 in TBS overnight at 4oC with gentle shaking. After washing once in ddH20, and two changes of TBS, blots were developed using the Amplified Alkaline Phosphatase Immuno-Blot Kit of Biorad (Goat Anti-rabbit, Cat. # 170-6412) which can detect as low as 10 pg on a blot.
Statistics
Comparisons between group means +/- standard error of the means were analyzed using t test software of GraphPad, and 2-sided P values are given. For the comparison of the commercial sex worker (CSW) cohort to normals or HIV-1 infected patients, we used the Welch’s two tailed t test for our analysis of differences in genomic copies of HERV-K102 pol since the variance in the proviral copies for the CSW was much higher than for controls or for the HIV-1 patients. A p value less than 0.05 was considered statistically significant.
RESULTS
Highly vacuolated cells developed when cord blood mononuclear cells (CB) were cultured in IMDM but not when cultured in RPMI (data not shown). Many of the large granular cells had the morphology of CD14++CD16+ more mature macrophage-like cells (Fig. 1A, H, E stain) similar to that reported by others when CB cells were cultured in IMDM [17]. Electron microscopy (EM) showed high levels of 100 nm immature particles accumulated in the vacuoles with Env spikes, and the high degree of vacuolation gave the mononuclear cells a foamy appearance as shown in Fig. (1B, C). It was noteworthy that no cell surface budding was found for the immature particles by EM (data not shown). We suspected an endogenous source of particles as a similar degree of vacuolation was observed when autologous serum or normal human serum replaced fetal calf serum (Supplemental Fig. 1). Of the human endogenous retroviruses, HERV-K HML-2 is the most recent and biologically active [1], so we used our own primer set for HERV-K HML-2 pol as described previously [3] in order to further characterize its replication and its association with foamy virus-like pro-perties.
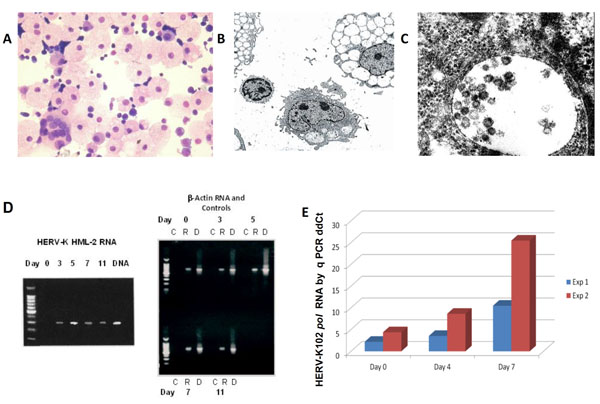
Characterization of HERV-K102 particle production and replication in vitro in CB. (A) H&E staining of day 7 CB cytospins showing the presence of highly vacuolated foamy macrophages [17] amongst normal small lymphocytes (400X). Note the presence of a rare multinucleated giant macrophage. (B) Electron microscopy of vacuolating CB cells day 11 at 1500X. (C) At 100,000X immature particles can be seen in the vacuoles which averaged about 100 nm. Env spikes are also noted. No cell surface budding was observed. (D) RT-PCR and PCR over an 11 day time course of CB cells cultured in 10% FCS-IMDM with our novel HML-2 primer set described previously [3]. Left side: HML-2 RT-PCR where the last lane is purified DNA. Right side: Control β-actin (lane marked C) without the addition of reverse transcriptase, β-actin RT-PCR lanes (lanes marked R) and DNA lanes (marked D). Representative of 6 or more experiments. (E) Real time quantitative RNA ddCt PCR using HERV-K102 pol primer sets over a 7 day CB culture [3]. Uracil-N –glycosylase (UNG) treated reverse transcribed RNA templates showed no signals for HERV-K102 and the 18 S RNA control indicating no contaminating genomic DNA was present (data not shown). Two experiments (Exp 1, Exp 2) on separate cord blood donors are plotted. Note that the induction at day 0 is thought to be due to delays in cord blood processing. The error bars are too small to plot.
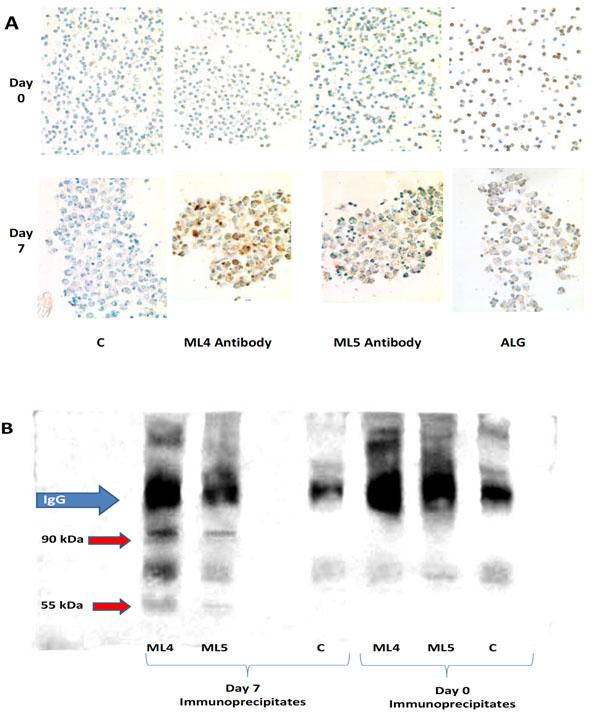
Expression of HERV-K102 Env with vacuolation. (A) By immunohistology performed with the affinity purified ML4 rabbit antibody to HML-2 type 1 Env and with the affinity purified ML5 rabbit antibody to HERV-K102 Env diluted 1/1000 [3]. Expression is detected only in vacuolating cells, and only on CB cultured for 7 days but not in uncultured day 0 CB. Purified positive control rabbit antilymphocyte globulin (ALG) and negative control rabbit globulin (C) are also shown. (B) Western blotting with affinity purified ML4 antibody diluted 1/1000 on ML4 and ML5 immunoprecipitated envelope proteins (precursor 90 kDa, processed 55 kDa) from day 7 cultured CB cells but not isolated from uncultured day 0 CB cells. C refers to control purified rabbit antibody (pre-serum).

Increased proviral copies of HERV-K102 pol may be associated with resistance to HIV-1 acquisition. DNA was extracted from plasma and the HERV-K102 pol ddCt ratio was performed with respect to 18S RNA as previously described [3] on UNG treated templates to ensure genomic DNA and not particle associated cDNA was assessed. Individual ddCt ratios were plotted: Light blue = 16 Canadian HIV-1 patients on antiretroviral therapy (HIV-1 T) with a mean genomic copy ratio of 1.07 +/- 0.14. Dark blue = 10 Canadian HIV-1 patients not on antiretroviral therapy (HIV-1 NT) with a mean of 1.16 +/- 0.21. Note there was no statistical difference in the genomic copy ratios of HERV-K102 pol between the Canadian HIV-1 infected patients whether or not they were on therapy and neither group alone or together was statistically different from normal. Pink = Commercial Sex Workers (CSW) who had shown at least 3 years of HIV-1 resistance to transmission despite daily exposures to HIV-1 [15,16] with a mean genomic copy ratio of 4.21 +/-0.73. This was significantly elevated above normal (0.88 +/-0.37; p<0.0005) and above the 2 standard error cutoff of 1.62 (N=30, data not shown).
A RT-PCR time course over 11 days with our novel primer sets for HERV-K HML-2 pol [3] is shown in Fig. (1D) (left panel, with b-actin and other controls on the right panel). HML-2 pol RNA was not detected at time zero, but progressively elevated, peaked at day 5 and was reduced on day 7. In this set of experiments on day 7, the RNA was harvested after cell lysis of about 30% of highly vacuolated cells. Thus, the levels of HML-2 pol RNA were lower on day 7 than day 5. This is consistent with preferential lysis of HML-2 RNA expressing cells on day 7 which related to high levels of vacuolation. RT-PCR products were isolated from the gels of day 5 CB cultures (Fig. 1D) and sent for sequencing. In 6 of 6 cases, for the 295 nts cDNA, only HERV-K102 pol sequences were obtained (AF164610 nts 4271 to 4565), although we did not formally rule out low levels of other HML-2 group members, nor at other time points.
We confirmed the HML-2 pol RT-PCR findings in Fig. (1D) related to HERV-K102 pol expression, by using our real time quantitative PCR (qPCR) HERV-K102 pol ddCt ratio method which employs 18s RNA as the comparer gene [3]. As seen in Fig. (1E), HERV-K102 pol containing RNA was remarkably induced over the 7 days of culture, with 10 fold or 25 fold induction of RNA by day 7. Here the cells were harvested before 30% of cells lysed and thus, the day 7 RNA was higher than day 4. It should be noted that the small induction at time zero (Fig. 1E) is thought to relate to a delay in the processing of the CB, as these particular neonates were delivered in the middle of the night. We did not detect HERV-K HML-2 pol RNA in CB nor adult peripheral blood mononuclear cell (PBMC) samples, which were freshly isolated (data not shown). The RNA signals obtained in Fig. (1E) were not due to contaminating genomic or particle associated DNA as DNAse treatment of the samples was performed with the QIAGEN RNA Mini kit with RNA clean up. As well, we performed parallel analysis on the same plates at the same time with Amperase-UNG in the Universal MasterMix, which digests the cDNA made during the reverse transcriptase step. This completely abolished both the detection of HERV-K102 and 18sRNA RNA signals (data not shown) as expected and showed no DNA signal was present. Furthermore, testing for DNA in these cultures demonstrated that HERV-K102 pol total DNA (cDNA and genomic) increased only 5.4 fold and integrated proviral copy number about 3.3 fold over the 7 days of culture (data not shown) again ruling out contamination by DNA for the results presented in Fig. (1E) performed on RNA. Thus, we confirmed the strong induction of HERV-K102 RNA was associated with particle production and vacuolation over the 7 days of culture.
Using our novel affinity purified ML4 and ML5 rabbit antibodies described previously to type 1 HML-2 (ML4) and HERV-K102 (ML5) Env peptides, respectively [3], the expression of Env in the vacuolated day 7 cells was demonstrated by immunohistology. Env was not present in uninduced freshly isolated CB (Fig. 2A), as expected. Non-vacuolated cells did not appear to express Env. Cell surface expression of Env was not detected by flow cytometry on induced CB or adult PBMCs when cultured in IMDM, although weak signals could be detected when cells were permeabilized (data not shown). This was compatible with our finding that no cell surface budding in normal immunocytes was detected by electron microscopy and that no membrane accentuation was found by immunohistology (Fig. 2A). Thus, normal mononuclear cells do not appear to express HERV-K102 Env at the cell surface, even in cells that are highly vacuolated and producing copious amounts of particles. Thus, in normal cells permissive for HERV-K102 particle production, cell surface expression of HERV-K102 Env was not found.
We next turned to western blots using the ML4 antibody to visualize antigens immunoprecipitated by the ML4 and ML5 antibodies. For the betaretroviral HML-2 group of endogenous retroviruses, the association of the transmem-brane unit with the surface unit (SU) is noncovalently associated and not attached through a disulphide bridge [1,9]. Also, FV require processed Env for particle production and infectivity [6,7, 14]. Thus, in order to show that the Env was processed, the western blot methodology employed non-reducing gels. Under non-reducing conditions then, specific bands of 90 and 55 kDa (Fig. 2B) representing intact and processed Env (respectively) were demonstrated as expected for HML-2 Env [9] for day 7, but were not detected in the day 0 harvests, the latter which do not express HERV-K102 pol RNA, nor exhibit vacuolation. The immunoblot in Fig. (2B) reveals the presence of purified primary rabbit IgGs used during the immunoprecipitation due to the second labeled (goat anti- rabbit) antibody used for the blot develop-ment. This conveniently allowed confirmation that the conditions used were not reducing and shows equivalent immunoprecipitated materials were added to all wells.
We then compiled a list of unique features of a foamy virus (FV) as exemplified by PFV, a chimpanzee FV originally mistaken as a human foamy retrovirus [6]. As shown in Supplemental Table 1 online, HERV-K102, a type 1 HML-2 [1,2] appeared to have many if not most of the hallmark features of PFV, the latter that is replication competent and fully infectious [4-8,14]. Most significantly this included DNA genomes in circulating particles, lack of a Rec-like domain, no immunosuppressive domain in the transmembrane region of Env, processed Env protein which is not covalently linked, particles which do not mature, no cell surface budding in normal cells, release by lysis in normal cells, massive induction of particles forming vacuoles giving cells a foamy appearance, start of the genome with TGTG, and so on. Importantly, FV have a reversed lifecycle to orthoretroviruses and genomes in particles are predominately DNA [4] that we showed previously for HERV-K102 particles in vivo [3].
In addition to characterizing the unknown foamy effect on CB, we also wanted to examine the potential role of HERV-K102 particles in HIV-1 infection. Nixon’s group had already shown compelling evidence that HML-2 antigens were expressed on the cells surface of HIV-1 infected cells but not normal cells, and that HML-2 specific antibodies and CD8 clones from an elite controller can eliminate HIV-1 infected cells in vitro [18-20]. The sequence recognized by the CD8 clones [18,19] was identical to an Env sequence found in HERV-K102. In addition, a number of groups have provided evidence of molecular interference of HIV-1 replication and infectivity by HML-2 proteins in vitro [21,22]. Thus, the expression of HML-2 proteins appears to be surrogate markers of HIV-1 infected cells and expression of proteins appears to be involved in a novel host defense system of humans. However, we wanted to address if the particles themselves might play a role in host defense. We were also curious about the finding that high particle production in the order of 106 to 1012 particles per ml of plasma was detected in some bloodborne pathogen infections, but only low HERV-K102 viremia in HIV-1 patients [3]. Drawing from our in vitro work which clearly showed the existence of permissive and nonpermissive conditions for HERV-K102 replication, we entertained the notion that HIV-1 transmission is associated with nonpermissive conditions, whereas in contrast, individuals chronically exposed to HIV-1 and other pathogens, but yet not infected, may have experienced permissive conditions for HERV-K102 replication. For this investigation, genomic DNA isolated from plasma was examined for evidence of HERV-K102 replication in a Nairobi commercial sex worker (CSW) cohort (n=14) known to have been resistant to HIV-1 transmission for at least 3 years [15,16] versus HIV-1 infected patients (n=26). Genomic DNA in plasma is thought to be derived from cells recently lysed in vivo, and thus could conceivably enrich for cells in which HERV-K102 replication may have been active but then underwent lysis. As shown in Fig. (3), remarkably, the mean genomic HERV-K102 pol DNA ddCt ratio as detected in plasma of the CSW cohort performed in the presence of uracil-N-glycosylase (UNG) to digest any cDNA templates present, was found to be almost 5 fold elevated over normal (4.21 +/-0.73; n=14 versus 0.88 +/- 0.37; n=30, p= 0.0005 Welch’s two tailed t test) not found in the North American HIV-1 patients irrespective of whether (T) or not (NT) they were on antiretroviral activity. Twelve of the 14 CSW had ddCt levels at 2.0 or greater (86%) while one had a ddCt ratio around 12. This is below the maximum integration reported for PFV [13]. Thus, the high level of HERV-K102 integration in the HESN female CSW cohort substantiates the potential importance of the high replication of HERV-K102 in the prevention of HIV-1 acquisition.
DISCUSSION
We found for the first time that the foamy appearance of CB is associated with a high level of replication of HERV-K102. The high levels of viral particles produced under permissive conditions in vitro is reminiscent of the high levels of HERV-K102 particles, approximately 1012 per ml of plasma, in some patients with bloodborne pathogen infections, except HIV-1 positive patients [3]. The rate of induction of HERV-K102 particles in vivo under permissive conditions was measured in a patient with chronic fatigue syndrome, achieving 2.55 x 1011 particles per ml of plasma over 84 hours and was confirmed to be cDNA by digestion with UNG (data not shown). We hypothesize that a rapid induction of this endogenous retrovirus in vivo may be relevant to combatting a formidable pathogen, like HIV-1.
The budding of retroviral particles into vacuoles is a property only ascribed to foamy retroviruses [5-7,14], therefore it may be possible to classify HERV-K102 as a replication competent human foamy virus. To corroborate its replication competence, significantly Env expression and processing [4-8] was demonstrated. The electron microscopy analysis demonstrated that Env studding was visible on the particles produced in foamy macrophages and is consistent with the proper processing of Env and possibly being infectious [14]. This is in direct contrast to Tera-1 cells, where HML-2 particles budding through the cell surface only infrequently showed Env studding [23]. Moreover no Env protein was detected by western blotting in Tera-1 cells, and not surprisingly, the particles bearing HML-2, 22q11 proviruses were not infectious [23]. Since a comparison of the genetic motifs and key attributes of HERV-K102 with PFV clearly showed HERV-K102 has the hallmark features of non-pathogenic FV, our work may be first to describe HERV-K102 as a foamy retrovirus specific to humans. The genetic stability of FV is unprecedented in the world of viruses which have an RNA intermediate [8]. Thus, in retrospect, in terms of stability, safety and nonpathogenicity, that a replication competent HML-2 active in vivo in modern day humans, would have DNA genomes and other salient features of FV, was not too surprising.
Visually, the cell types producing particles appeared to be foamy macrophages and T cells, and studies by flow cytometry confirmed that the highly granulated cells expressed CD14 or CD3 but not CD19 as expected (data not shown). It may be significant that these are the cell types also susceptible to HIV-1 infection [24]. This is also consistent with the possibility that HERV-K102 particle production could directly protect against HIV-1 replication. In culture, it appeared that vacuolation initiated in a small percentage of cells visible by day 2, and then progressively spread to or was induced in the rest of the culture, visually estimated to be about 30% of the cells (data not shown). In preliminary experiments, we found CD14 cell depletion by magnetic beads completely abrogated vacuolation over 7 days (data not shown) suggesting that HERV-K102 particle production might initiate in activated macrophages or alternatively, that macrophages were needed for HERV-K102 particle initiation. Clearly it will be important to discern between these two possibilities as it may have repercussions for our understanding of the phenomenon of HIV-1 transmitted/founder (T/F) strains, which generally use the CCR5 co-receptor [24]. That HIV-1 exists as macrophage tropic and/or T cell tropic strains was first observed by Gartner et al., in 1986, and in this seminal paper they were first to propose HIV-1 likely gains entry into the host through the infection of macrophages [25]. This is consistent with our finding that the majority of HERV-K102 particles seem to be produced in foamy macrophages and as developed below, may be protective against HIV-1 acquisition.
It is now well established that most T/F strains use the CCR5 co-receptor [24,26] but it remains to be determined whether macrophages are the initial targets in HIV-1 acquisition or not [24-27]. Resolving this issue might have been confounded by the indiscriminate use of non-permissive versus permissive culture conditions for HERV-K102 particle production. For example the original observations for the macrophage tropic and T cell tropic strains were both performed in RPMI [25] which, we have found is non-permissive for HERV-K102 particle production (data not shown). However, in a more recent article which casted doubt on the requirement for initial macrophage infection to successfully establish transmission [24], RPMI was used for studying T cells but DMEM was used for the culture of monocyte derived macrophages (MDM). The former would be non-permissive for HERV-K102 particle production and would likely give HIV-1 a replication advantage, while the latter would be permissive, and would interfere with HIV-1 replication. Consequently, this difference may in part explain why it was reported HIV-1 T/F strains replicated at about 2 to 3 logs lower levels in MDM [24] in conflict with the original findings by Gartner et al., 1986 [25]. Accordingly, to better understand T/F strains and their significance, a more physiological determination of the target cell type of HIV-1 transmission in vitro might be to use CB cultured in IMDM as demonstrated here under conditions permissive for HERV-K102 particle production. Similarly, interpretation of HIV-1 T/F strains, based on in vivo simian models also may not be appropriate as they lack HERV-K102 [1]. Instead, humanized mice models might be a better choice to examine this important issue of target cell types of T/F strains in vivo and what limits the transmission of HIV-1 [28].
The finding that the induction of HERV-K102 particle production in macrophages leads to foamy macrophages and that particle production and/or release might be blocked in HIV-1 patients [3], may also be of some additional clinical significance. Foamy macrophages are known to initiate atherosclerosis, and persons infected with HIV-1 are known to be at higher cardiovascular risks [29]. Our in vitro work seems to imply that instead of high cholesterol causing atherosclerosis, it may more frequently be due to HERV-K102 particle production in response to viral infections [3]. Since the particles and the vacuoles in which they bud are all lipid bilayers involving cholesterol, this might help explain why these cells have higher cholesterol and how cholesterol streaks accumulate in plaques [30]. Thus, the novel charac-terization of a replication competent human foamy virus, HERV-K102 reported here, may have broader repercussions not only for protection against infectious diseases [3,9-1,18-22], and tumors [31,32], but for cardiovascular disease risks [29,30]. Presumably foamy macrophages persist when HERV-K102 particles are induced but not released through lysis.
The high level of integrated HERV-K102 proviruses found in the HESN cohort is consistent with high levels of HERV-K102 particle production. In contrast, the HIV-1 infected group was unable to produce and/or release HERV-K102 particles at the same rate as the HESN cohort [3] (Fig. 3). Thus, it seems HIV-1 acquisition may have been blocked by high HERV-K102 particle production. The foamy macro-phages producing HERV-K102 particles are morphologi-cally distinct and resemble CD14++ CD16+ macrophages previously described by Stec et al., 2007 which were obtained from cord blood cells also cultured in IMDM [17]. In HIV-1 patients, it is the CD16+ monocytes in blood which harbor HIV-1 DNA and that are more permissive to HIV-1 infection [33]. Moreover CD14++ CD16+ monocytes express the highest level of CCR5 [33] consistent with the notion that HERV-K102 particle induction is likely induced in the same cells preferentially infected by HIV-1. That HIV-1 induces HERV-K102 particle production in vivo has been shown by two groups [3,9] and more recently the induction of HERV-K102 expression in vitro by HIV-1 has been demonstrated [34]. Thus, HIV-1 likely confronts HERV-K102 particles when first entering the host. Finally, it has been recently shown that in chronic HIV-1 infection the CD16+ monocytes remain activated irrespective of antiretroviral therapy [35] also consistent with low HERV-K102 particle production in chronic HIV-1 infection [3]. While the gatekeeper of HIV-1 transmission is yet to be elucidated [26], HERV-K102 activation and/or particle production may be early defence mechanisms helping to limit HIV-1 replication and due to ongoing activation, may also contribute to the maintenance of the viral set point.
Accumulating evidence strongly implies HML-2 activity may be protective such as against HIV-1. For example, T cell responses to HML-2 antigens including a type 1, HML-2 Env epitope which behave as surrogate markers of HIV-1 infected cells, have been linked with protection against HIV-1 as has been demonstrated in an elite controller. These T cells clear HIV-1 and other lentivirus infected cells in vitro [18,19]. As well, antibodies to HML-2 Env transmembrane regions were found at higher levels in HIV-1 patients (titer of 1350) vs HIV negative controls (titer of 450) and eliminate HIV-1 infected cells in vitro by an ADCC mechanism [20,36]. However, here titers were higher in aviremic HIV-1 patients on ART than in elite controllers; while all human samples were positive. In contrast, we previously reported that HERV-K HML-2 type 1 Env specific (ML4 peptide) and HERV-K102 Env specific (ML5 peptide) antibodies were demonstrated in 80% and 70% of HIV-1 patients, respectively, but which were only rarely demonstrated in normal adults [3]. As well, such antibodies were also detected in patients with other bloodborne pathogens, but at lower frequency. Curiously, antibodies to the ML4 and ML5 peptide sequences of HERV-K102 Env surface unit were not reported in the Michaud et al. studies as they did not examine “type 1” HML-2 specific peptide sequences [36]. However, it seems the latter may be important as antibodies to HERV-K102 type 1 envelope protein were recently found to mediate apoptosis of breast cancer cell lines in vitro and in vivo by signaling through the p53 pathway and involved increased activity of caspase 3, 8 and 9 [31]. Thus, type 1 HERV-K102 HML-2 Env antigen cell surface expression on tumor or virally transformed cells may not be just a beacon, but remarkably, may itself be a mediator of cell killing.
The existence of T and B cell responses to HML-2 anti-gens generally seems paradoxical as these are self-antigens and ordinarily there should be immunological tolerance to these antigens. This is a concern as several groups are now proposing surrogate vaccines for tumors [31,32] and HIV-1 [18-20,36] based on HML-2 immunogens. The notion of T helper cell independent B cell responses has been well established in immunology, and it is known multivalent large antigens can provide the initial triggering, whereas intra-cellular Toll-like receptors (TLRs) and other pathogen recognition receptors can provide signal 2 to expand the response [37,38]. Interestingly the involvement of endogenous retroviruses has also been recently implicated in T independent B cell responses [37]. It is tempting to speculate that Env antigen on HERV-K102 particles may trigger T-independent B and possibly T cell responses, whereas once inside the cells, the HERV-K102 DNA genomes may activate TLRs for signal 2. This may help explain why there was a notable differential between normals and HIV-1 patients for HERV-K102 Env specific epitope antibodies [3] but antibodies to HML-2 transmem-brane epitopes were scored positive in all human sera tested [36]. Thus, it may be that HERV-K102 particles could be used to stimulate a surrogate innate immunity vaccine against HIV-1, but not HML-2 antigens.
Our study has limitations. We observed a five-fold increased level of HERV-K102 integration in the HESN over normals which was not found for individuals already infected with HIV-1 and is consistent with the notion that in vitro, under permissive conditions, integrations increased about 3.3 fold over 7 days (data not shown). This suggests the HESN women experienced past high levels of replication as evidenced by the abnormally high levels of integration over normal controls. The question remains however, as to whether inherited genetic polymorphisms in the African Nairobi cohort might alternatively explain the higher integration level of HERV-K102, not found in the North American HIV-1 positive groups, which were mostly male. While one could address genetic polymorphisms in female residents of Nairobi not exposed to HIV-1, such samples were not available to us. Even if they were, and shown to be normal, the question would remain as to whether the Nairobi, HESN cohort some of whom are related, acquired or inherited the extra integrated copies of HERV-K102. Unfortunately, the participants in the Nairobi cohort were all females and access to germ cells for testing this, would not be very practical. The finding of variable copy number in the cohort which ranged from normal up to 12 might argue that it was not likely due to inherited genetic polymorphisms where a more consistent pattern might have instead been expected. On the other hand, it is clear that the protection against HIV-1 transmission in the Nairobi cohort was not genetically determined in that women who were absent from the sex trade for about 6 to 12 months, were at a much high risk of becoming HIV-1 infected upon returning to work [16]. Indeed, in the Nairobi, Kenya HESN cohort, an unknown innate immunity mechanism presumably provoked by repeated exposures to pathogens and involving interferon responses, appeared to protect against HIV-1 transmission [reviewed in 39, 40]. Moreover, that a state of protection is also associated with adaptive immune quiescence [39] would be consistent with our findings that PHA and IL-2 supplementation in the IMDM cultures blocked HERV-K102 particle production (data not shown). The contribution of repeated exposures presumably to other pathogens, is reminiscent of the findings reported in injection drug use HESN, where it has been shown protection related to the sharing of needles and the stimulation of innate immunity [41] and is consistent with our work showing various viruses stimulate HERV-K102 particle production in vivo [3]. Interestingly, more recent evidence in the Nairobi cohort has shown Mx2 expression, which inhibits HIV-1 integration, was associated with medroxyprogesterone use and the latter was associated with protection against HIV-1 transmission [42]. Remarkably, the stimulation of HERV-K HML-2 genome expression by progesterone has been observed after estradiol treatment in a human breast cancer cell line [43] suggesting HERV-K102 particle production may have been also enhanced in the Nairobi HESN cohort associated with Depo-Provera use. Altogether, these findings provide a compelling stance that increased integration of HERV-K102 in the Nairobi HESN cohort, was likely acquired rather than inherited. As a final argument, while HERV-K HML-2 polymorphisms have been studied in human populations around the world, variances in HERV-K102 proviral copy number have not been found [44].
In summary, this study provides further evidence of the replication competence of HERV-K102 particles both in vitro and in vivo and substantiates not only that HERV-K102 has salient features of foamy retroviruses, but that under permissive conditions high level virion induction may occur in vitro and in vivo. This report provides the first initial evidence that high levels of HERV-K102 particles might be associated with early protection against HIV-1 acquisition. A major cell type producing HERV-K102 particles and/or in which particle production initiates, appears to be the macrophage and thus, may be intimately relevant to HIV-1 transmission by CCR5 co-receptor strains. Whether or not increased HERV-K102 integration levels are found in other HESN cohorts clearly needs to be examined, particularly those involving males where inherited versus acquired provirus copy number can be more easily assessed. As well, HERV-K102 particle production and/or antibody responses should also be investigated in elite controllers and compared with non-controllers. These and other future efforts elucidating the role of HERV-K102 particles in limiting HIV-1 replication might be expected to help advance HIV-1 functional cure research. Finally, since HERV-K102 appears to be a non-pathogenic foamy virus of humans, may be protective and capable of high particle production, and is induced by HIV-1 in vivo and in vitro, this work raises the possibility that HERV-K102 particles could be exploited for prevention vaccines and/or the sterilizing cure of HIV-1 infected patients, alone or as a replication competent vector to transfect specific inhibitors of HIV-1 replication.
CONCLUSION
This work raises the possibility that high HERV-K102 particle production in macrophages may serve to help limit HIV-1 replication and possibly transmission. Whether HERV-K102 particle production is the long-awaited, scientific paradigm shift instructive for the development of an HIV-1 vaccine and/or functional cure [45], remains to be determined.
Authors’ Contributions
MPL designed, arranged and directly managed the execution of the work, including ethics approval, obtained a grant for Applied Biosystems Inc., real time PCR equipment purchase, wrote the manuscript and performed statistical analysis. LL designed the HML-2 unique primers, carried out much of the earlier lab work, and helped to write the manuscript. AG, and FD-M provided operational support and funding, direction, constructive criticism of the manuscript and expertise. All participated in and approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We would like to acknowledge the technical assistance of Yangxun Hou, Deana Bellfoy and Ioana Moldovan. A special thanks to Louise Pelletier for the immunohistological preparations and staining. We would like to also thank Drs. Keith Fowke and Francis A Plummer for access to the CSW HESN samples, for which we are very grateful. Many thanks to Katherine Laderoute for editorial assistance.
Funding
Supported by the Blood Safety Program at Health Canada and the Public Health Agency of Canada and by an Innovative Research Grant from the Office of the Chief Scientist at Health Canada. In kind contributions were made by the Research Institute at the Children’s Hospital of Eastern Ontario and by the Department of Pathology and Laboratory Medicine at the University of Ottawa.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Web site along with the published article.
Additional file 1: Supplemental Fig. ( S1). Vacuolation does not depend upon source of serum used for culture. Supplementary Table S1. Comparison of features of Prototypic Foamy Virus to Type 1 HERV-K (HML-2) HERV-K102.

