All published articles of this journal are available on ScienceDirect.

Immune Suppression by Myeloid Cells in HIV Infection: New Targets for Immunotherapy
Authors Info & Affiliations
Abstract
Over thirty years of extensive research has not yet solved the complexity of HIV pathogenesis leading to a continued need for a successful cure. Recent immunotherapy-based approaches are aimed at controlling the infection by reverting immune dysfunction. Comparatively less appreciated than the role of T cells in the context of HIV infection, the myeloid cells including macrophages monocytes, dendritic cells (DCs) and neutrophils contribute significantly to immune dysfunction. Host restriction factors are cellular proteins expressed in these cells which are circumvented by HIV. Guided by the recent literature, the role of myeloid cells in HIV infection will be discussed highlighting potential targets for immunotherapy. HIV infection, which is mainly characterized by CD4 T cell dysfunction, also manifests in a vicious cycle of events comprising of inflammation and immune activation. Targeting the interaction of programmed death-1 (PD-1), an important regulator of T cell function; with PD-L1 expressed mainly on myeloid cells could bring promising results. Macrophage functional polarization from pro-inflammatory M1 to anti-inflammatory M2 and vice versa has significant implications in viral pathogenesis. Neutrophils, recently discovered low density granular cells, myeloid derived suppressor cells (MDSCs) and yolk sac macrophages provide new avenues of research on HIV pathogenesis and persistence. Recent evidence has also shown significant implications of neutrophil extracellular traps (NETs), antimicrobial peptides and opsonizing antibodies. Further studies aimed to understand and modify myeloid cell restriction mechanisms have the potential to contribute in the future development of more effective anti-HIV interventions that may pave the way to viral eradication.
INTRODUCTION
The human immunodeficiency virus (HIV) pandemic is responsible for over 35 million cases and 1.5 million deaths a year [1, 2] with a huge economic impact on resource-poor settings. The most frequent mode of HIV-transmission is through sexual contact leading to persistent infection involving multiple components of the immune system [3]. The viral suppression by antiretroviral therapy (ART) improves health, increases survival in HIV-infected patients and reduces the rates of transmission of the virus. However, current therapeutic regimens do not eradicate the virus completely and are unable to restore health in HIV-infectd patients who also become prone to co-morbidities, such as increased cardiovascular disease, bone disorders and cognitive impairment [4]. In the light of available data, there is a dire need to develop curative strategies such as immune-therapeutic approaches to address the public health burden posed by HIV [5]. Further studies aimed at understanding HIV pathogenesis may provide missing links for the development of interventions towards a successful cure.
Following HIV entry through mucosal surfaces of the genital or gastrointestinal (GI) tract, viral translocation occurs across the mucosal epithelium leading to interactions with CD4 T cells and macrophages, accounting for the major aspects of HIV pathogenesis (3). After years of research on CD4 T and other lymphoid cells, recent works point to the emergence of the myeloid cells as central players in HIV pathogenesis [6-9]. This review summarizes the contribution of myeloid cells on immune dysfunction in HIV infection, including the vicious cycle of immune activation driven by myeloid cells and its effect on the patient outcome. The role of macrophages, monocytes, dendritic cells (DCs) and neutrophils in the context of HIV infection will be discussed highlighting the target mechanisms for potential new interventions.
MYELOID CELLS IN HEALTH AND PATHOLOGY OF HIV INFECTION
Myeloid and lymphoid progenitor cells are the two major cell lineages of the immune system originating from common hematopoietic stem cells in the bone marrow and giving rise to the myeloid cells (monocytes, macrophages, DCs, neutrophils and others) and lymphoid cells (T cells, B cells, natural killer (NK) cells and others) [10]. These cells make up the cellular components of the innate and adaptive immune systems. Monocytes, macrophages, DCs and neutrophils are the phagocytic cells that express pattern recognition receptors (PRRs) to recognize molecular patterns found on pathogens (pathogen associated molecular patterns, PAMPS) [11]. Toll-like receptors (TLRs) are PRRs that recognize distinct PAMPs and play a critical role in innate immune responses. They participate in the first line of defense against invading pathogens and contribute significantly in inflammation, immune cell regulation, survival, and proliferation [12]. HIV RNA, on the other hand, encodes for multiple PAMPs that can be recognized by TLR7 expressed in plasmacytoid dendritic cells (pDCs) and TLR8 expressed in both monocytes and myeloid dendritic cells (mDCs) [13].
Host innate and adaptive immune responses are employed in early HIV infection to defend against the viral invasion [14]. Before systemic viral progression and as a part of innate immune response at the site of infection, DCs and monocytes secrete type 1 interferons and induce the expression of interleukin (IL)-15 (a pleiotropic and multifunctional cytokine essential for NK cell proliferation and activation and T cell homeostasis) [15]. The subsequent inflammatory signals generated by these cells contribute to the development of a specific acquired immune response.
Macrophages along with DCs represent a crucial subset in HIV infection due to their influence on viral transmission, target cell infection and presentation of HIV antigens [16]. These antigen presenting cells (APCs) link innate with adaptive immunity by activating T cells. Besides promoting transmission and spread, the monocytes, macrophages, and DCs also play a pivotal role by serving as viral reservoirs [6]. Neutrophils, traditionally considered as essential effector cells of the innate immune system, are now also implicated in regulation of the adaptive immune system as they co-localize and actively communicate with T cells [8]. Moreover, reduction in neutrophil counts (neutropenia) is predictive of bacterial and fungal infections in AIDS patients. All these myeloid cells can be distinguished from each other by a number of cell surface markers listed in Table 1 along with the main characteristics and functions of these cells in health and in HIV pathogenesis. (Table 1)
Characteristics of cells of myeloid lineage in health and in the context of HIV infection.
| Cell Type | Cellular Markers | Primary Locations | Average Life Span | Functions/Features | Interaction with HIV | References |
|---|---|---|---|---|---|---|
| Macrophages | CD68+ EMR1+ CD14 |
Mucosal surface Tissues Brain (Microglia) Lungs (Alveolar) Liver (Kupffer cells) Bone (Osteoclasts) |
Week to Months | Phagocytose cellular debris and pathogens Antigen presentation Arginine production under stimulation M1/M2 polarization Tissue remodelling |
Restricted HIV replication Resist cytopathic effects Neuropathogenesis Induce PD-1 expression Induce IL-10 production Major viral reservoir Activate CTL by cross-presentation of HIV antigens |
[54, 56, 68, 71, 115] |
| Monocytes | CD14hiCD16-CD14+CD16+ CD14lo/CD16hi |
Peripheral blood | Few days | Patrol blood for surveillance Precursor to macrophages Precursor to DCs Interact with LPS via CD14 Phagocytosis |
Restricted HIV replication Resist cytopathic effects Neuropathogenesis Induce PD-1 Induce IL-10 CD16+ is more permissive Viral reservoir |
[16, 50, 54, 56, 78] |
| Myeloid DCs | Lin−HLA-DR+ CD11c+ CD1c+ CD123− |
Peripheral blood Cerebrospinal fluid |
Week to Months | Antigen presentation Arginine production under stimulation Phagocytosis Induce T cells in cell-mediated immunity |
Less productive viral replication Transfer in cis and trans Potential to use in vaccines Induce PD-1 Induce T cell IL-10 production |
[81, 88, 91, 93, 128] |
| Neutrophils | CD15 CD16 CD62L CD33 CD13 CD11b CD11c CD24 |
Peripheral blood | Less than 2 days Or Several days when activated |
Most abundant leukocytes Arrive first at an injury site Promote acute inflammation Modulate T-cell responses Regulate humoral response Induce M1/M2 macrophages Kill bacteria via NETs Produce antimicrobial peptides Phagocytosis |
Infection not reported Binding through FC-g NETs inactivate HIV HIV induces DCs to produce IL-10 to counteract NETs. Production of a-defensins |
[8, 105, 110] |
| Myeloid derived suppressor cells (MDSC) | HLA-DR- CD11b+ CD33+ CD14+ CD15- CD66b CD11b VEGFR1 CD62lo CD16lo |
Peripheral blood | Few days | Prevent damage by acute and chronic inflammation. Impair T cell function via ROS, arginase-1 and cytokines Suppress IFN-g production Phagocytosis |
Increase with plasma viral load Increase with CD4+ decrease HIV infection or exposure to HIV-encoded protein Tat could drive MDSC generation in vitro |
[8, 120, 121] |
| Low density Granulocytes (LDGs) |
CD15 CD33 CD13 CD11c CD24 CD16hi CD62Llo |
Peripheral blood | Few days | Impair T cell function via ROS, arginase-1 and cytokines Induce PD-L1 on neutrophils Phagocytosis Decreased adhesion |
Increase with plasma viral load Increase with CD4+ decrease Inhibit T-cell proliferation |
[8, 111, 112] |
Selected ongoing clinical trials using immunotherapeutic approaches.
| Group | Intervention | Main Targets Cells and/or Mechanisms | Study Phase | Study Population | Title | Trial ID No. |
|---|---|---|---|---|---|---|
| Antibodies | Anti PD-L1 Ab BMS-936559 |
Myeloid cells | Phase I | HIV-infected patients on Suppressive cART | Safety and immune response of BMS-936559 in HIV-infected people | NCT02028403 |
| UB-421 antibody | Monocytes and T-lymphocytes |
Phase II | Asymptomatic, treatment-naive, HIV seropositive Patients, aged 18 + |
Study to evaluate safety and efficacy of UB-421 antibody | NCT01668043 | |
| Immuno-metabolic agents | Atorvastatin | Monocytes | Phase IV Pilot Study |
HIV patients, n~15, aged 18 + | Modulation of monocyte activation by atorvastatin in HIV infection | NCT01263938 |
| vitamin D and phenylbutyrate | Myeloid Cells | Phase II | Treatment-naive HIV-infected patients, aged 18 to 75 years | Immunotherapy to induce important antimicrobial defence mechanisms and decreased inflammation. | NCT01702974 | |
| DC based vaccines | Arcelis™ AGS-004 | Immunogenicity | Phase I/II | acute and chronic HIV patients suppressed on ART | Study to Evaluate the Immunologic Response and Virologic Impact of AGS-004 | NCT02042248 |
| Inflammatory and other molecules | Chloroquin | Autophagy inhibitor | Phase III | HIV-infected patients on ART, aged 18+, Undetectable viral load |
Daily trimethoprim-sulfamethoxazole or weekly chloroquine among adults on ART in Blantyre, Malawi | NCT01650558 |
| Probiotics | Microbial translocation & immune reconstitution | Phase II | Patients with chronic HIV infection and undetectable viral load | Treatment with probiotics and its role in bacterial translocation and immune reconstitution in HIV infection. | NCT01908049 | |
| Aspirin | Immune activation | Phase II | HIV-infected patients on continuous ART for 48+ weeks | Modulation of immune activation by aspirin | NCT02155985 |
Source: www.ClinicalTrials.gov (accessed on August 18, 2014).
HIV INFECTION AND INNATE IMMUNE DYSFUNCTION
The key factor causing immune dysfunction in HIV infection is immune activation leading to viral replication and a gradual decrease in CD4 T cells attributed to the combination of enhanced destruction and reduced thymic production [17]. Their destruction is linked to direct infection and bystander effects of syncitia formation with macrophages, immune activation and immune senescence (aging of the immune system) [18]. A recent study by Doitsh G et al. has shown that non-productive HIV infection activating caspase-1-dependent pyroptosis is a major inflammatory pathway of programmed cell death in resting CD4 T cells [19]. Initially pyroptosis was described as a program of cell death induced by intracellular bacteria in macrophages and characterized by the release of cytokines such as IL-1β (an endogenous cytokine activated by caspase-1 that induces leukocyte tissue migration and coordinates inflammation by the expression of multiple other cytokines), IL-18 (a cytokine that activates T cells and macrophages and stimulates the production of interferon gamma, IFN-γ), IL-6 (A pro inflammatory cytokine associated with HIV disease progression) and tumour necrosis factor alpha (TNF-α, a pleiotropic proinflammatory cytokine involved in multiple biological processes such as cell proliferation, differentiation, apoptosis, lipid metabolism, and coagulation) [20, 21]. Pyroptosis directly links CD4 T cell depletion with chronic inflammation by releasing cytoplasmic contents and pro-inflammatory cytokines including IL-1β [19].
The peripheral blood depletion of CD4 T cells evident in early HIV infection is reverted close to normal cell counts thereafter. Later on with disease progression, the CD4 T cells decrease slowly by about 50-100 cells per μL per year [22]. The most important damage on T-cell homoeostasis happens in fact very early in the GI tract where a massive depletion of activated CD4 T cells expressing chemokine receptor 5 (CCR5) occurs [23]. In addition to loss of total CD4 T cells, profound changes in T-cell subsets happen, including preferential loss of T-helper-17 (Th17) cells and mucosal-associated invariant T cells, which are important for defence against bacteria [24]. The profound depletion of Th17 cells in the GI tract, together with enterocyte apoptosis and enhanced GI tract permeability, leads to an increased release of microbial products such as lipopolysaccharides (LPS) into the circulation [25]. Finally, in the lymphoid tissue the destruction of the fibroblastic reticular cell network, collagen deposition and restricted access to the T-cell survival factor IL-7 further contributes to depletion of CD4 naive T cells [26].
IMMUNE ACTIVATION AS A VICIOUS CYCLE DRIVEN BY MYELOID CELLS
HIV infection is characterised by a marked increase in immune activation, which involves both the adaptive and innate immune systems along with abnormalities in blood coagulation [27]. The drivers for immune activation include the following: 1) the direct effects of HIV as a ligand for endosomal TLR7 and TLR8 leading to over-production of interferon alpha (IFN-α) [28]; 2) increased microbial translocation, with LPS as a potent activator of cell surface TLR4 leading to the production of pro-inflammatory cytokines such as IL-6 and TNF-α [29]; 3) co-infection with other viruses such as cytomegalovirus (CMV) that induce profound expansion of activated CMV specific T cells and a reduced ratio of Th17 and regulatory T cells (Tregs), especially in the GI tract [25]. Even in patients on long-term ART with adequate CD4 T cell restoration evidence of residual inflammation persists [30]. Markers of residual inflammation in ART treated patients have been significantly associated with mortality, cardiovascular disease, cancer, neurological disorders and liver dysfunction [31, 32]. Furthermore, one study found low-levels of HIV replication which may contribute to persistent inflammation as ART including raltegravir in virally suppressed HIV-infected patients was associated with a decrease of immune activation in some patients [33]. This study supports the conclusion that residual viral replication drives in part the immune activation and that raltegravir intensification can reduce the extent of immune activation by further suppressing residual viral replication. The production of inflammatory cytokines, such as IL-6 by monocytes predicted serious non-AIDS associated events and death, which were not associated with CD4 or CD8 T cell activation in long-term ART treated patients [9]. The depletion of CD4 T cells initially in the GALT and the persistent systemic immune activation, represent the driving forces to immune suppression leading to AIDS [34].
Microbial translocation and biomarkers associated with monocyte activation (sCD14) and coagulation (D-dimer) have also been identified as independent predictors of disease progression and mortality in HIV infection [35-37]. During intestinal inflammation, resident monocytes recruit neutrophils by producing chemoattractant gradient. Upon reaching the inflammatory site, neutrophils selectively release chemoattractants to attract macrophages for a second-wave inflammatory response that ensues for the next several days [38]. Administration of ART can only partially repair HIV-inflicted gut mucosal injury [39]. A study our group contributed to has recently shown that a 12 week IL-7 adjunctive therapy in ART suppressed HIV-infected patients led to increases of both CD4 and CD8 T-cells in peripheral blood and an improvement in gut barrier integrity as measured by decreased neutrophil infiltration and decreased TNF-α levels in the rectosigmoid lamina propria [40]. Globally, the persistence of myeloid-driven immune activation on long-term ART has been independently associated with elevated frequency of non-AIDS events that mimic age-related complications [5].
Amino acid starvation is increasingly considered as a potent immunoregulatory mechanism. For example, reduction of tryptophan (Trp) levels, by the enzymatic activity of indoleamine 2,3-dioxygenase (IDO) or Tryptophan 2,3-dioxegenase (TDO) in the kynurenine (Kyn) pathway suppresses immune responses in cancers [41] and chronic viral infections [42]. Macrophages and DCs when stimulated by IFN-γ, cytotoxic T-lymphocyte antigen-4 (CTLA-4, an inhibitory receptor that suppresses T cell activation) and TLRs, also produce intracellular IDO [42, 43]. We recently examined Trp catabolism and its impact on Th17/Treg balance in a large cohort of HIV-infected patients grouped into ART-naïve, successfully treated, and elite controllers [44]. It was found that in ART-naïve patients, increased IDO levels were associated with Treg expansion and an altered Th17/Treg balance, which were normalized under ART.
PERSISTENCE OF HIV ACTIVATION FURTHER CONTRIBUTES TO T CELL DYSFUNCTION
Persistence of immune activation in both aging and in HIV infection has been linked to the loss of optimal T cell responses characterized by the impairment of cytokine secretion and CD8 T cell cytotoxicity. Polyfunctional T-cells correspond to cells simultaneously capable of producing cytokines and chemokines such as IL-2, TNF-α, IFN-γ, macrophage inflammatory protein-1β (MIP-1β) and CD107a (a marker of granulation) as well as displaying cytolytic functions [45]. In HIV infection, optimal polyfunctional memory T-cell responses are rapidly lost during the chronic phase as T-cells become increasingly dysfunctional. This dysfunctional phenotype is considered as “exhaustion” and correlates with the level of immune activation [46]. Factors promoting this T-cell exhaustion include persistence and high levels of antigenic stimulation, DC inhibition and upregulation of inhibitory receptors such as Programmed Death-1 (PD-1) on CD4 and CD8 T cells [47]. The T cell function is considered to be regulated by the expression of PD-1, which can be blocked by binding with PD-L1 mainly expressed by monocytes, macrophages and dendritic cells, thus impairing T cell proliferation and cytokine production [48-50]. HIV, on the other hand, induces T cell expression of both PD-1 and CD57, a marker of replicative immune senescence and clonal exhaustion [48, 49]. The monocytes and macrophages, by interacting with HIV and up-regulating PD-L1, are now considered as central players in the shutdown of T cell response via PD-1 on CD4, CD8 T cells and B cells [8, 50]. Recently, PD-L1 was also reported to be expressed on the surface of neutrophils, another member of myeloid lineage [8] which further compromises T cell function in HIV infection. Both innate and adaptive immune responses depend significantly on the activities of myeloid cells including macrophages, monocytes, dendritic cells and neutrophils.
MACROPHAGES PLASTICITY AND POLARI-ZATION IN HIV INFECTION
Macrophage infection by HIV has been reported since the discovery of the virus in the mid-1980s by Gartner et al. [51] and Ho et al. [52]. Macrophages play a key role at the initial contact between the virus and the host when translocation of transmitted founder HIV happens across the mucosal epithelium, the largest reservoir of macrophages in the body [3]. On these mucosal surfaces, HIV encounters potential target cells mainly in the lymphocyte and macrophage populations of the lamina propria. In contrast to CD4 T cells, the function of macrophages in genital and gut mucosal tissues remains less studied due to methodological difficulties of sampling and isolation of these cells from tissues [53].
Macrophages are highly plastic, dynamic and terminally differentiated cells that act at the first line of defence by directly clearing the pathogen and cell debris through phagocytosis and by directing other leukocytes [54]. They also function as antigen presenting cells (APCs) by presenting processed pathogen antigen peptides to the CD4 T cells via MHC II pathway [55]. It is widely reported that the alveolar, intestinal, and adipose tissue macrophages exhibit what is essentially immunoregulatory functions, whereas Kupffer cells, peritoneal and secondary lymphoid tissue macrophages generally kill microbial invaders [56, 57].
Recent evidence indicates that macrophages, apart from their classic phagocytic functions, ensure critical roles in tissue development by promoting angiogenesis and vascular remodeling in postnatal organs [58]. “Primitive hematopoiesis”, originating from unique yolk sac-derived progenitor cells, takes place before definitive hematopoiesis and generates erythrocytes and macrophages that migrate through the yolk sac vasculature to colonize the fetus [59, 60]. These cells persist and maintain the macrophage pool into adulthood, without being superseded by adult bone marrow-derived or blood monocyte-derived cells [61-63]. Yolk sac-derived primitive macrophages contribute to adult cell types in particular for Langerhans cells and microglia, the tissue macrophages of the brain [63]. Microglia represent the only non-neuroepithelial cells found in the parenchyma of the central nervous system (CNS) and solely originate from the yolk sac and migrate into the CNS during mid-embryogenesis. Microglia are distinctive from any other myeloid cells as they are maintained independently of any contribution from the blood and do not require hematopoietic stem cells (HSCs) as a source of replacement for senescent cells. Microglial functions are dissimilar from those of blood-derived monocytes, which invade the CNS only under pathological conditions [64]. Yolk sac-derived macrophages from mice are different from HSC-derived macrophages as they express F4/80bright and CD11bdim, and are independent of myeloblastosis oncogene (Myb) and FMS-like tyrosine kinase 3 (Flt3), whereas HSC-derived macrophages are F4/80dim, CD11bbright, and Myb- and Flt3-dependent [65]. The existence of such “self-maintained” macrophages in tissues have been confirmed in humans and have not been studied in the context of infections. This cell type represents new avenues of research that could have important implications in HIV neuropathogenesis and persistence.
Macrophage polarization depicts the functional heterogeneity of macrophages, which mirrors Th1-Th2 polarization, where the M1 type represents pro-inflammatory macrophages and M2 represents anti-inflammatory macrophages [56]. The M1/M2 polarization ratio reflects the bidirectional macrophage-lymphocyte interaction: Th1 cells drive M1 polarization via IFN-γ whereas Th2 cells direct M2 polarization via IL-4 and IL-13 [56]. M1 macrophages produce reactive oxygen species (ROS) and express high levels of pro-inflammatory cytokines, including IL-1β, IL-12 (a cytokine that acts mainly on naïve CD4 T cells and induces antiviral cytotoxic T lymphocyte response), IL-23 (a cytokine that acts mainly on memory CD4 T cells and stimulates IFN-γ production via the transcription activator STAT4), IL-18 and TNF-α. IL-18 has also been reported to be elevated in HIV infection [66]. An M1 phenotype is associated with acute viral infections including HIV and leads to the production of cytokines that drive viral replication and tissue damage [67]. In vitro studies have shown M1 cells to inhibit HIV infection, whereas M2 cells inhibit viral replication at a post-integration stage [68]. HIV endocytosis causes productive infection in macrophages in M1 as compared to resting macrophages. These cells were also found to have an increased rate of HIV-1 endocytosis as compared to M2 cells [69]. Macrophages also express high levels of C-type lectins such as mannose receptor that can facilitate macrophage-mediated transmission of HIV to CD4 T cells [67].
Besides resting memory CD4 T cells, macrophages represent an important cellular reservoir for HIV [70]. Macrophages are widely recognized as the earliest target of HIV with subsequent infection of T cells. Similar to monocytes, macrophages are resistant to the cytopathic effects of the virus and better able to evade the defensive action of the immune system in HIV infection [71]. Therefore, the infected macrophages keep on producing and harbouring the HIV for a longer period. They are able to cross the blood-tissue barriers and can transfer HIV to all tissues and organs. It has also been reported that the macrophages isolated from patients receiving ART harbour replication competent HIV [72]. These findings indicate that the infected macrophage, are of critical importance in HIV pathogenesis contributing to early-stage cell-to-cell viral transmission, dissemination and persistence throughout the body. The current literature clearly demonstrates the importance of HIV interactions with macrophages throughout the course of infection and stresses the need for interventions to block these interactions for a successful cure/prevention.
CIRCULATING MONOCYTES AND HIV INFECTION: AN UNDERRATED INFLAMMATION SOURCE
HIV infection of monocyte/macrophage lineage cells has long been recognized to contribute to disease pathogenesis [51, 52]. Monocytes are targets of HIV infection as they express the CD4 receptor and chemokine co-receptors for HIV entry. However, both in vivo and in vitro data suggest infection of circulating monocytes with HIV to be rare in comparison to CD4 T cell compartments [72-74], which is often latent and could lead to productive infection during differentiation into macrophages. Later on, sophisticated approaches using in situ hybridization coupled with simultaneous surface immuno-phenotyping revealed a higher level of monocyte infection and demonstrated the production of viral mRNA in these cells indicating productive infection [75, 76]. At any given time throughout the course of infection, less than 1% of monocytes harbour HIV [77].
HIV restriction mechanisms in monocytes or myeloid cells have been recently identified and include enrichment of host restriction factors, such as tetherin, apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G/F (APOBEC3G/F) [78] and SAM domain and HD domain-containing protein 1 (SAMHD1) [79]. SAMHD1 is one of the important myeloid cell restriction factors that has an RNase activity and is responsible for preventing HIV infection by directly degrading the HIV-1 RNA [80]. These restriction factors, mostly induced by cytokines such as INF-α, exert potent antiviral activity in cell culture. However, HIV infection progresses efficiently due to the potent viral antagonists called as accessory proteins, namely Nef, Vif, Vpr and Vpu. Vif counteracts the cytidine APOBEC3G, Vpu and Nef deactivate the antiviral factor Tetherin besides dowregulating CD4 and the SIV counterpart of Vpr, the Vpx protein, antagonizes SAMHD1 [71, 79, 80].
Monocytes are the phagocytic immune cells in blood, representing 10-30% of all circulating mononuclear leukocytes. Monocyte life span comprises of a few days in the bloodstream and when activated or differentiated into macrophages or DCs, their life-span is prolonged for up to several months [71, 81]. These cells bridge innate and adaptive immunity as precursors of APCs, the macrophages and DCs [54]. This cell heterogeneity likely supports specialization for different types of invading pathogens. The vast majority of circulating monocytes express CD14, a receptor for LPS-binding protein, which is considered as a primary monocyte marker. We previously demonstrated monocyte activation profiles and their links with macrophage polarization using clinical samples from patients with Q fever which is caused by infection with Coxiella burnetii, an obligate intracellular bacterium that establishes infection by highjacking the host immune response [57].
Monocytes are able to resist the HIV-induced cytopathic effects, and therefore persist throughout the course of infection disseminating the virus to tissues. They serve as an HIV reservoir, which prevents viral eradication in HIV-infected patients [70]. Recovery of infectious virions from monocytes isolated from patients on ART suggests their contribution to viral reservoirs [72]. However, the mechanism of HIV latency in these cells remains open to further exploration. Monocyte differentiation into macrophages renders these cells increasingly susceptible to HIV infection and permissive to viral gene expression and production of infectious viruses [82]. Interestingly, the differentiation of monocytes into macrophages stimulates HIV production in the infected monocytes, suggesting their role in both viral latency and reactivation [82, 83].
PD-1 over-expression on monocytes is reported to be associated with an increase in microbial products and IL-10 in the plasma of HIV-infected patients. This study also reported the PD-L1 triggering of PD-1-induced IL-10 production in monocytes that leads to reversible CD4 T cell dysfunction which is described as a new function for PD-1 [50]. Monocytes along with macrophages are also linked with HIV-induced dementia and play an important role in HIV-associated neuropathogenesis [84]. The entry of HIV infected monocytes into the CNS is dependent on impaired inflammation as it was shown in a mouse infection model that the TAT viral protein upregulated the expression of inflammatory cytokines and adhesion molecules such as monocyte chemoattractant protein-1 (MCP-1/CCL2, a chemokine which specifically mediates monocyte chemotaxis), TNF-α, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1) and facilitated the entry of monocytes into the brain [85]. Furthermore, HIV infection causes impairment of monocyte functions including phagocytosis, intracellular killing, chemotaxis and cytokine production. Functional and phenotypical differences are observed within monocytes in the peripheral blood [83], as there exist at least two monocyte populations: CD14-positive (CD14++ CD16-) and CD16+ positive (CD14+CD16+) subtypes [86]. The later represent about 5% of the monocytes in circulation and are known to play an important role in bacterial infections as they are more pro-inflammatory. Identification of HIV-induced dysfunction in different monocyte subsets will provide new directions for appropriate interventions aimed at restoring functions of these cells.
MYELOID DENDRITIC CELLS (mDCs) AND HIV: THE DAMAGED HEART OF THE VIROLOGICAL SYNAPSE
HIV infection of DCs was first reported in 1987 [87]. These sentinel cells that are present at mucosal surfaces are involved in the transfer of HIV to CD4 T cells [88]. In vitro studies have shown CD4 T cells becoming infected when co-cultured with HIV-pulsed DCs and have also revealed the potential mechanisms, which facilitate DC-mediated HIV transmission to CD4 T cells in a biphasic mode as: 1) HIV capture and release by DCs; and 2) Productive HIV infection of the DC. These potential mechanism include trans or early transfer mode, in which the virus is rapidly relocated from endosomal compartments in DCs to the DC/T-cell contact zone via a virologic synapse [89]. In this mode, C-type lectin receptor DC-SIGN on DCs interacts with gp120 viral envelope protein to capture and transmit HIV to CD4 T cells which results in enhanced viral replication [89]. A productive infection of DCs leads to the cis or late transfer mode where progeny virus is transferred to CD4 T cells [90]. DC immune-receptor (DCIR) mediated transmission of HIV by both cis and trans infection pathways has also been reported [89].
DCs are a diverse set of cells that coordinate innate and adaptive immune responses. DCs are a rare population of WBCs in blood or tissues which can be divided into subsets based on expression of cell-surface markers, anatomical distribution and immunological function. mDCs and pDCs in the blood and langerhans cells in the tissues make up the major DC subsets. Among total PBMCs in blood, mDCs and pDCs represent 0.5-2% of cells [81].
mDCs are characterized by their ability to secrete high levels of IL-12, whereas pDCs can prime antiviral adaptive immune responses by producing high levels of type 1 interferons [88]. mDCs differentiate from monocytes in blood under specific cytokine milieu and are important APCs that patrol submucosal surfaces in search of foreign invaders [91]. DCs link the innate with adaptive immune response by endocytosis and subsequent lysis of invading pathogens in the endolysosome or proteasome, leading to presentation of epitopes to T cells in association with MHC I and II [81, 91, 92]. All DC subsets express CD4 and the co-receptors for HIV chemokine co-receptors CCR5 and CXCR4 rendering them susceptible to infection with HIV [88]. However, HIV replication in DCs compared to CD4 T cells is generally less productive, and the frequency of infected DCs in vivo is often 10- to 100-fold lower [88]. The mechanism used for restriction of productive viral infection could be attributed to lower expression of CD4, CCR5 and CXCR4, or internalization of HIV leading to rapid and extensive degradation. Viral infectiveness could also be modulated by expression of host restriction factors, including the antiretroviral proteins SAMHD1 and APOBEC3G, which may represent an important innate immune mechanism against retroviral infection [6].
Our group has previously analysed the DC populations in a cohort of well-characterized HIV-infected patients with different rates of disease progression and reported alterations in circulating mDC counts and phenotypes during the very early stage of infection that persist after the apparent control of HIV replication with ART [93]. These alterations can be used as an early criterion of disease progression in HIV-infected individuals and should be targeted in the design of therapies involving autologous mDC vaccination [93].
An important mechanism in viral pathogenesis of HIV is the impairment of the antigen-presenting function of DCs [88]. Viral protein Nef-mediated down regulation of the co-stimulatory molecules CD80 and CD86 in HIV-infected DCs may also be responsible for impairment in T cell functions [94]. HIV infected DCs enhance T-cell production of IL-10 which further causes immunosuppression [95]. Globally, these results suggest that productive HIV infection of mDCs undermines the direct induction of T-cell-mediated immunity.
It has been shown that productive HIV replication occurs in mDCs for up to 45 days [96]. These cells may survive longer within the lymph nodes due to cytokines present in the microenvironment, contributing to the spread of HIV trans infection and persistence of viral reservoirs [88].
NEUTROPHILS AND HIV INFECTION: NEW PARTNERS IN CRIME
HIV does not infect neutrophils but reduction in the neurophil count (neutropenia) is a frequent complication of advanced HIV infection [97]. Neutropenia is a significant risk factor for bacterial and fungal infections in AIDS patients. In addition to decreased cell counts, a variety of qualitative defects in neutrophil function have been described, including abnormal chemotaxis, phagocytosis, oxidative metabolism, and bacterial killing [98]. Neutrophils from HIV-infected patients produced IL-12 constitutively [99] which could contribute to chronic immune activation.
Neutrophils also referred to as polymorphonuclear granulocytes, are in a majority among all leukocytes [100]. Initially at the infection site, neutrophils rapidly accumulate and actively communicate with T cells and later migrate to the draining lymph nodes to induce and regulate cellular and humoral immune responses. Neutrophils specifically produce ROS, such as hydrogen peroxide, and arginase (ARG)-1 to impair the T cell functions [8]. Recently, increased expression of PD-L1 on neutrophils has also been reported in HIV infection and tuberculosis [8, 101]. Such findings are of importance as these cells are much more numerous than DCs and macrophages in tissues and may further contribute to the T cell exhaustion.
Neutrophil production of antimicrobial peptides, such as α-defensins, indolicidin and lactoferrin also has implications in the control of HIV infection [102-104]. α defensins specifically down regulate the expression of CD4 and CXCR4 and block viral entry to the target cell [102]. The secretion of these peptides in the breast milk of HIV patients prevents viral transmission to the child and is associated with chronic immune activation [105]. Neutrophils link innate with adaptive immunity by means of α-defensins as these peptides induce antigen specific T cell functions and are able to recruit naive T cells and DCs at the site of injury [106]. Moreover, these peptides are reported to induce CD4 and CD8 T cells to produce IFN-γ and perforin [107].
FC-γ receptors present on the surface of neutrophils can bind HIV tagged with specific anti HIV opsonizing antibodies resulting in HIV internalization through phagocytosis [108-110]. HIV-Neutrophil binding may also be responsible for the spread of the virus as it has been shown in vitro that exposure to inflammatory stimuli increases the neutrophil binding levels with HIV by at least twofold and further leading to increased infection of lymphocytes with neutrophil bound HIV as compared to free HIV [109]. Neutrophil interactions with HIV have therefore both beneficial and harmful implications for the immune system as well as transmission of infection as described above and reviewed previously [110].
In an inactivate state, neutrophils life span is shortened and the dying neutrophils undergo necrosis or apoptosis [110], however, when activated by the invading pathogens, they live longer in order to provide their immunological functions [100]. Neutrophils plasticity and phenotypical changes have been reported by a number of studies [98, 111, 112]. The emerging data points out the fact that the neutrophils are not an end-stage uniform cell population but in fact they develop distinct phenotypes and/or subsets in response to a wide range of physiological (e.g. growing age) and pathological (e.g. inflammation and infection) conditions as reviewed recently [113]. In fact, LPS is able to induce differential subsets of neutrophils in the circulation [100, 111], which may be explained by different states of neutrophil maturation rather than the existence of distinct cell lineages [100].
Tsuda et al. have reported existence of two more neutrophil types as compared to normal neutrophils. The first type they described are the neutrophils with a pro-inflammatory phenotype expressing IL-12 and CC3 which were isolated from mice resistant to methicillin-resistant staphylococcus aureus (MRSA). The second type they described are the neutrophils isolated from MRSA-susceptible mice with an anti-inflammatory phenotype expressing IL-4, IL-10 and CCL2 [114]. Whether these pro-inflammatory and anti-inflammatory neutrophils play a role in the induction of classical M1 or alternate M2 macrophages remains to be elucidated [56, 115]. Neutrophils ability to be both pro-inflammatory and anti-inflammatory cells that regulate the cellular and humoral immunity highlights their contribution in adaptive immune response.
Inflammatory and antibacterial activities of neutrophils mediated by the release of ROS are also known to cause cell death through recently discovered “neutrophil extracellular traps” (NETs), which is a collection of their nuclear contents [108, 116]. Similar to neutrophils, NETs are also reported to possess antimicrobial proteins including neutrophils elastase (NE), myeloperoxidase (MPO), lactoferrin, and gelatinase, which could enable NETs to kill pathogenic invaders in a highly efficient manner with minimal host damage [108, 117].
NETs have been described in many studies as webs to trap pathogens including bacteria, fungi, viruses and protozoan parasites [118]. Saitoh et al. have recently reported NETs to trap and inactivate HIV [119]. TLR7 and TLR8 receptors on neutrophils can bind with viral nuleic acid causing ROS production, which promotes NET formation and subsequently leads to the elimination of HIV [108, 119]. Interestingly, HIV counteracts NETs by inducing DCs to produce IL-10. These recent study findings highlight the important contribution of neutrophils on HIV pathogenesis. Neutrophil functions in the context of HIV infection may have a previously unforeseen role for in HIV pathogenesis. Therefore, future studies deciphering HIV-neutrophil interactions have potential to contribute in designing strategies for an HIV cure.
MYELOID DERIVED SUPPRESSOR CELLS (MDSCs) AND LOW DENSITY GRANULOCYTES (LDGs): THE NEW KIDS IN THE BLOCK
Myeloid derived suppressor cells (MDSCs) have been recently described to be circulating in peripheral blood as a part of the inflammatory response in a variety of pathologies of multiple origins [8, 120]. HIV disease progression was strongly correlated with the level of MDSCs which were functionally suppressive to T cell responses through induction of arginase (ARG)1 by direct cell contact [121]. MDSCs represent a novel mechanism of T cell dysfunction to prevent potential tissue damage induced by inflammation in HIV-infection, sepsis and cancer [8, 121, 122]. These cells likely originate from activated polymorphonuclear cells and cause impairment of T cell function by production of ROS, ARG-1, cytokines and induction of Tregs [122]. MDSCs have been widely reported in cancer; however, studies on their roles in viral infections are less well defined [121, 123, 124].
A recent study has reported that the ARG expressing cells among PBMCs of HIV-infected patients were low-density granulocytes (LDGs) exhibiting a similar morphology to normal-density granulocyte [112]. This study also revealed that the increased frequencies of LDGs correlated with disease severity highlighting the important role of these cells in HIV pathogenesis (Table 1). LDGs in this study were considered as activated neutrophils that have been degranulated owing to increased cell surface expression of CD66b, CD63 and CD11b and decreased intracellular ARG 1 expression [112]. Importantly, a recent study reported that the presence of LDNs in the blood of HIV patients correlated with the upregulated expression of PD-L1 on neutrophils [8]. Globally, these novel findings clearly indicate that neutrophils and related cells play a more important role in HIV infection than initially thought and represent new avenues of research.
MYELOID CELLS AND THEIR ASSOCIATED IMMUNE FUNCTIONS AS IMMUNOTHERAPEUTIC TARGETS
As discussed myeloid cells play an important role in the vicious cycle of HIV associated generalised chronic inflammation mainly due to release of inflammatory cytokines and modulation of PD-1 pathway which causes immune dysfunction by supressing T cells (Fig. 1). And yet proven therapies targeting them are lacking. Here we discuss the ongoing exploration of immunotherapy targeting myeloid cells, such as inhibitors of PD-1 pathway. Myeloid cells overexpress PD-L1 which interacts with T cells via PD-1 leading to immunosuppression in HIV infected patients [8, 47]. In a tumour model of lymphopenic mice, anti-PD-L1 antibody administration demonstrated increased activation and persistence of adoptively transferred T cells, including a higher number of CD8 T cells [125]. Use of antibodies in vitro for blocking PD-L1-PD-1 interactions in PBMCs from HIV-infected persons and PD-1 blockade in vivo in a SIV-infected macaque model have reported promising results [48, 126]. The first study reported that in cells isolated from HIV infected treatment-naïve patients, the blockade of the pathway augmented HIV-specific CD4 and CD8 T-cell function [48]. Moreover, in the second study using a macaque HIV infection model, PD-1 blockade by anti PD-1 antibody was well tolerated and resulted in rapid expansion of virus-specific CD8 T cells with improved functional quality [126]. There is at least one open phase-I clinical trial aimed to determine safety, pharmacokinetics and immunotherapeutic activity of an anti-PD-L1 antibody (BMS-936559) in HIV-infected patients on suppressive cART (Table 2). Therefore, the blocking of PD-L1 signaling in vivo by immunotherapeutic approaches could have beneficial effects on HIV-infected patients.
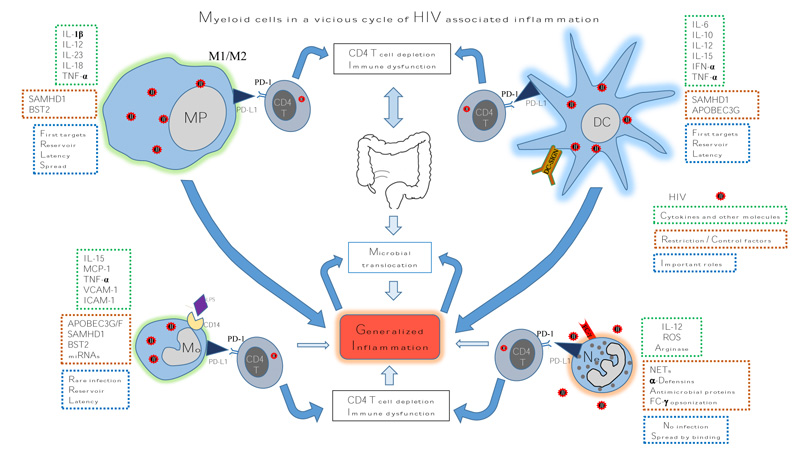
Myeloid cells in a vicious cycle of HIV associated inflammation. Macrophages, monocytes, DCs and neutrophils play an important role in HIV pathogenesis by various mechanisms leading to generalized inflammation. Myeloid cell up regulation of PD-L1 enhances their interaction with CD4 T cells via PD-1 which further results in CD4 T cell depletion and immune dysfunction. Expression of restriction and/or control factors by these cells is circumvented by HIV. Modulation of production of cytokines and other molecules also contributes to the inflammation.
Therapeutic immunization by different approaches such as administration of autologous monocyte-derived DCs (MD-DCs) carrying a variety of antigens ex vivo could reduce the need for lifelong ART. In a rhesus monkey model of SIV infection, it was first demonstrated that an effective and durable SIV-specific humoral and cellular immunity can be elicited by vaccination with DCs pulsed with inactivated SIV as the source of viral antigens [127]. However, not much has been done on therapeutic immunization in HIV infection and the results of few clinical studies conducted in HIV-infected adults have been summarised by Garcia et al [128]. These DC based vaccines enhance antigen-specific T-cell responses and induce potent protective immunity in experimental murine models and in human patients with cancer and chronic infections [128]. Autologous MD-DCs have also been used in human trials. An investigational immunotherapeutic product (AGS-004, Argos Therapeutics Inc., NC, USA) that consists of autologous dendritic cells co-electroporated with in vitro transcribed RNA encoding four of the patient’s own HIV antigens is being evaluated in a phase I/II study (Table 2) based on the encouraging immunogenicity and tolerance observed in a Phase I study [129, 130]. A recent proof of concept randomized trial on HIV patients reported safety, tolerability, and immunogenicity associated with a control of viral replication using autologous MD-DCs pulsed with autologous heat-inactivated whole HIV [131]. A significant decrease in plasma viral load by more than one log and a consistent increase in HIV specific T cell responses in immunized recipients were observed. Such positive findings strongly support further investigations of new candidates and optimized strategies of therapeutic immunization [131].
Macrophages, like DCs, are a heterogeneous cell population which are major targets of HIV infection. They are functionally polarized into pro-inflammatory (M1) and alternatively activated (M2) macrophages. The M1/M2 macrophage functional transition is achieved in vitro by the use of cytokines such as IFN-γ, TNF-α, IL-4 and IL-13 [56, 67]. A recent study explored the extent to which agonists triggering the TLR family affect HIV replication in polarized macrophages [7]. It was reported that a number of TLR agonists blocked HIV replication substantially when given before infection. The mechanisms of how TLR agonists exert their inhibitory action were also examined which has advanced our understanding of which and how TLR agonists block HIV infection in polarized macrophages and may facilitate the design of novel immunotherapeutic approaches [7]. Therefore, macrophage polarization is clearly implicated in the control of HIV infection in vitro at entry and post entry levels and further in vitro and in vivo studies have the potential of guiding research towards containment of infection or a possible cure.
Chloroquine (CQ) is a cheap and widely available antimalarial drug, which modulates variety of inflammatory cytokines making it a logical choice to be used as an immune-modulatory agent in several autoimmune disorders. The cellular endosomal pH has also been shown to be increased through CQ treatment which can lower IL-6 synthesis [132] suppressing HIV production from chronically infected T-cells and monocyte cell-lines [133]. CQ and its hydroxyl analogue hydroxychloroquine (HCQ) are reported to inhibit the in vitro replication of HIV [134] which highlights their potential as an immunotherapeutic agent to be used in conjunction with other antiretroviral medications. In one clinical trial, HIV-infected patients randomly assigning HCQ 800 mg/d or placebo for 8 weeks reported decreases in viral load [135]. Another study also reported a decrease in serum p24 levels and a decrease in IL-6 and immunoglobulin G levels [136] but both studies did not find any effect of the intervention on CD4 T cell counts. A recent prospective study by our group evaluated the effect of CQ on immune activation and CD4 T-cell recovery in immune nonresponding patients receiving successful ART [137]. Our results show that CQ was well tolerated but a 24 week treatment did not improve CD4 T-cell recovery, lymphoid and myeloid immune activation or inflammatory markers [137]. A separate phase III clinical trial is underway to assess the long-term benefits of CQ and trimethoprim-sulfamethoxazole (TS) prophylaxis on survival and disease control among HIV-infected patients who have viral load suppression and a good clinical response on ART (Table 2).
In addition to clinical trials that are already in process, there are several other targets which may prove to be useful in future clinical trials. For example, HIV-infected patients that resist disease progression for years, referred to as long term nonprogressors were shown to harbour an altered lipid metabolism defined by higher cholesterol production in their DCs and thus, therapies aimed at modulating cholesterol metabolism could have implications in the control of HIV infection [138]. Host restriction factors could also play a role in future treatments. SAMHD1 has emerged as a myeloid-cell-specific HIV restriction factor to be considered for future therapeutic opportunities. Enhancement of SAMHD1 function may help hosts develop potent innate and adaptive immune responses to HIV. Lastly, strategies aimed at in vivo induction of neutrophils extracellular traps (NETs) and stimulation of neutrophils to produce antimicrobial peptides could inhibit HIV infection. A list of recent ongoing clinical trials in various phases is shown in Table 2.
CONCLUDING REMARKS
This review has highlighted the multifaceted role of myeloid-lineage cells including macrophages, monocytes, DCs and neutrophils, in HIV infection. Multiple myeloid-derived cells are capable of restricting infection by multiple mechanisms, yet these same cells are also known to upregulate PD-L1 leading to immune dysfunction. Due to their impact in innate and adaptive immune response, various possible target mechanisms exist in these cells such as blockade of PD-1/PD-L1 interactions through anti PD-L1 antibodies, enhancement of macrophage polarization and NETs. Myeloid cell-derived treatments could provide rewarding and encouraging advancements for a successful cure through immunotherapy. Further investigations of the mechanisms underlying myeloid cell HIV restriction could not only lead to cure, but will certainly shed light on the innate immune response against retroviruses and aid in the future development of more effective anti-HIV interventions.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The authors acknowledge Ms. Angie Massicotte for coordination and assistance. This work was supported by the Canadian Institutes of Health Research (grant MOP #103230 and CTN #257), and Fonds de la Recherche Québec-Santé (FRQ-S): Thérapie cellulaire and Réseau SIDA/Maladies infectieuses, Québec, Canada. Dr. M.A. Jenabian was supported by a CANFAR/CTN Postdoctoral Fellowship Award. Dr. J.P. Routy is a holder of Louis Lowenstein Chair in Hematology & Oncology, McGill University.

