All published articles of this journal are available on ScienceDirect.

Immune Responses of HIV-1 Tat Transgenic Mice to Mycobacterium Tuberculosis W-Beijing SA161
Authors Info & Affiliations
Abstract
Background:
Mycobacterium tuberculosis remains among the leading causes of death from an infectious agent in the world and exacerbates disease caused by the human immunodeficiency virus (HIV). HIV infected individuals are prone to lung infections by a variety of microbial pathogens, including M. tuberculosis. While the destruction of the adaptive immune response by HIV is well understood, the actual pathogenesis of tuberculosis in co-infected individuals remains unclear. Tat is an HIV protein essential for efficient viral gene transcription, is secreted from infected cells, and is known to influence a variety of host inflammatory responses. We hypothesize Tat contributes to pathophysiological changes in the lung microenvironment, resulting in impaired host immune responses to infection by M. tuberculosis.
Results:
Herein, we show transgenic mice that express Tat by lung alveolar cells are more susceptible than non-transgenic control littermates to a low-dose aerosol infection of M. tuberculosis W-Beijing SA161. Survival assays demonstrate accelerated mortality rates of the Tat transgenic mice compared to non-transgenics. Tat transgenic mice also showed poorly organized lung granulomata-like lesions. Analysis of the host immune response using quantitative RT-PCR, flow cytometry for surface markers, and intracellular cytokine staining showed increased expression of pro-inflammatory cytokines in the lungs, increased numbers of cells expressing ICAM1, increased numbers of CD4+CD25+Foxp3+ T regulatory cells, and IL-4 producing CD4+ T cells in the Tat transgenics compared to infected non-tg mice.
Conclusions:
Our data show quantitative differences in the inflammatory response to the SA161 clinical isolate of M. tuberculosis W-Beijing between Tat transgenic and non-transgenic mice, suggesting Tat contributes to the pathogenesis of tuberculosis.
INTRODUCTION
Mycobacterium tuberculosis (M.tb) remains among the leading causes of death from an infectious agent in the world and exacerbates disease caused by the human immunodeficiency virus (HIV) [1]. It is estimated that one-third to one-half of the 30 million AIDS deaths can be attributed to tuberculosis [2]. Approximately 28% of individuals living with HIV and tuberculosis reside in South Africa [3]. In these endemic regions, the footprint of co-infection is striking and hampers disease control. Numerous groups have examined the influence of M.tb on the development of HIV disease [4-6] as well as the converse - the impact of HIV on tuberculosis progression [6, 7]. An individual with HIV is more likely to reactivate latent M.tb infection and progress to active tuberculosis [8-10]. AIDS patients co-infected with M.tb often present with atypical radiographic findings and disseminated disease [11]. At present, dissection of the relationship between these two pathogens remains challenging.
M.tb strains of the W-Beijing family have been associated with multiple global outbreaks and may be coupled to the HIV epidemic. Cowley et al. [12] genotyped M.tb strains from archived post-mortem specimens from the Western Cape region of South Africa and found W-Beijing strains were rare or absent from samples collected between 1930 through the mid 1960s, but strains were recovered from the 1996-2005 samples. Cowley suggests the emergence of these strains is coincident with the spread of HIV infection. Middelkoop et al. and Caws et al. also reported an association between W-Beijing strains and HIV infection in South Africa [3] and Vietnam [13], respectively. In addition, W-Beijing outbreaks can involve multi-drug resistant (MDR) organisms [14]; some are “hypervirulent” compared to other virulent laboratory strains because infected mice fail to induce protective TH1 responses needed to control bacterial replication, resulting in shortened survival [15]. Together, these reports suggest W-Beijing strains are found worldwide and are commonly associated with HIV-infected individuals.
While mice are used in tuberculosis research, this model is not utilized to study HIV because the virus does not replicate in this species. Whether any of the known viral proteins directly impact the host response to tuberculosis, independently of CD4 depletion, remains poorly defined. Tat, an HIV protein, is necessary and contributes to the transactivation of viral genes as well as some cellular genes [16-18]. Tat is released by infected cells, taken up by uninfected bystander cells [19], and is known to affect several host immune responses in vitro [20-24]. Our group and others have shown that Tat stimulates the expression of intercellular adhesion molecule 1 (ICAM1), endothelial selectin (E-selectin), and vascular cell adhesion molecule 1 (VCAM1) [21, 25, 26]. Still, other reports indicate Tat inhibits interferon-induced nitric oxide synthase gene activity in macrophages [27], which may impact the ability of the host to defend against infection with M.tb.
Since Tat is one of the best-studied HIV proteins and mediates many of the virus’s pathogenic effects, a transgenic mouse that expresses the Tat protein was used to study its effects in host defense against M.tb. Our laboratory has previously characterized the phenotype of the Tat transgenic (Tat-tg) mice [28] used in the present report. In this model, the Tat gene is under the control of the surfactant protein C (SPC) promoter that is active in type II alveolar epithelial and some bronchial cells of the lungs. We confirmed Tat expression by immunohistochemistry, reverse transcriptase quantitative PCR and by immunoblot analyses in lungs, but not heart or liver of transgenic mice. At baseline, the Tat-tg mice demonstrate notable phenotypic differences compared to the non-tg mice including “increased pulmonary cellular infiltration (but no visible differences in lung architecture), increased nitrotyrosine and carbonyl protein modifications, and increased levels of NFκB p65, manganese superoxide dismutase (MnSOD), and thioredoxin interacting protein” [28]. Although the Tat-tg mice showed increased indices of oxidative stress, there were no obvious signs of decreased viability, weight fluctuations, or accelerated aging compared to the non-tg animals. Also in the initial description of these mice, we found no significant changes in IL-1β, IL-12, or IL-6 at baseline, but the level of these inflammatory cytokines increase upon challenge of the Tat-tg mice with a sub-lethal dose of bacterial lipopolysaccharide (LPS).
To explore the role of HIV Tat on host susceptibility to M.tb, we infected Tat-tg and non-tg mice with a low-dose aerosol of a highly virulent W-Beijing strain of M.tb known as SA161 [29]. The pathogenesis of this strain has not been evaluated in this particular animal model. We found mortality was accelerated in the Tat-tg SA161 infected mice and disrupted granulomatous and inflammatory responses. We conclude this murine model is useful to address the basic pathogenesis of co-infection.
MATERIALS AND METHODOLOGY
HIV Tat-Tg Mice
The description of the plasmid construct, polymerase chain reaction (PCR), generation of the transgenic mice, and protein expression of Tat in the lung is provided by Cota-Gomez et al. [28]. Briefly, HIV-1 Tat 86 (86 amino acids), under control of the surfactant protein C (SPC)-promoter, is expressed by lung type II alveolar epithelial and bronchial club cells. Expression of the tat gene was quantified by quantitative PCR and the presence of Tat protein confirmed by immunohistochemistry.
Mycobacteria Infections
Mycobacterium tuberculosis W-Beijing SA161 (kindly provided by Dr. K. Eisenach) were grown in 7H9 broth containing 0.05% Tween-80 and stored at -70°C; M.tb H37Rv was similarly grown. Mice were used at 12 weeks of age. For infections, thawed aliquots of frozen bacterial cultures were diluted and placed into a nebulizer in a Middlebrook Airborne Infection Apparatus, calibrated to deliver approximately 100 bacilli into the lungs. The bacterial load was determined on days 7, 15, and 30 post-infection by plating serial dilutions of whole lung homogenates onto 7H11 agar. Colonies were counted after three-weeks of incubation at 37°C.
Ethical Statement
All animal experimental protocols were approved by the University of Colorado Denver and Colorado State University Animal Care & Use Committees (protocol #40901907(06)1E). All animal infection experiments were performed in a Bio-safety Level 3 animal facility, according to the regulations of Colorado State University.
Mouse Survival Assay
Survival times for M.tb H37Rv or W-Beijing SA161 infected mice were determined by observing the animals on a daily basis. Animal behavior and weight were monitored weekly and when a sustained drop in weight was observed over several days, the animals were euthanized [30].
Histopathology
Lungs from uninfected Tat-tg mice were collected and placed in 4% paraformaldehyde/phosphate buffered saline (PBS) for fixation and stained with hematoxylin and eosin (H&E) for histopathological analysis. The accessory lung lobe from Tat-tg and non-tg mice infected with M.tb W-Beijing SA161 was collected for histopathological analysis on day 30-post infection. The tissue was placed in 4% paraformaldehyde/phosphate buffered saline (PBS) for fixation and the sections stained with hematoxylin and eosin (H&E) [31]. The lung lesion areas were quantified relative to normal tissue area using a stereology based method referred to as the Area Fraction Fractionator with the investigator blinded to all treatment groups. The lung lesion area was determined on representative H&E stained sections. The area of inflammation relative to the normal tissue parenchyma was estimated from representative lung sections evaluated at 20X. A total of 8 to 12 fields were selected randomly by the computer and a counting frame (2000 µm2) containing probe points with a grid spacing of 200 µm, was used to define the areas of interest. The data are expressed as the mean percentage of lung affected by lesions of all the animals within a treatment group (n=4). Lesions represented by photomicrographs represent the section that is closest to the mean value in each group.
Quantitative RT-PCR of Inflammatory Mediators
Lung tissues from M.tb W-Beijing SA161-infected mice were homogenized in Trizol (Qiagen) and frozen at each time point. Total RNA was extracted and cleaned using Qiagen columns. mRNA was measured by real-time quantitative RT-PCR using ABI Prism 7900 Sequence detector at the University of Colorado Cancer Core. PCR primers (TNFα, IFNγ, and ICAM1) and probes were purchased from Applied Biosystems. Reverse transcription was performed at 48°C for 30 minutes followed by activation of TaqGold (95°C, 10 minutes). Experiments were performed in duplicates for each data point. Each PCR reaction included the standard curve (10 fold serially diluted mouse control RNA), test samples, no-template, and no-RT controls. Quantities of targets in test samples were normalized to the corresponding 18S rRNA.
Flow Cytometry for Surface Markers and Intracellular Cytokines
Mice were euthanized at the appropriate time points after infection at Colorado State University under BSL3 conditions. The lung was cleared of blood by perfusion through the pulmonary artery with PBS containing heparin (Sigma). The lungs were aseptically removed, teased apart, and treated with deoxyribonuclease IV (DNAse) (Sigma) and collagenase XI (Sigma) (45 minutes, 37°C). To obtain a single-cell suspension, the organs were passed through 40µm strainers (Becton Dickinson). The remaining erythrocytes were lysed and washed with Dulbecco’s modified Eagle’s minimal essential medium. Total cell numbers were determined by flow cytometry using BD™ Liquid Counting Beads (BD PharMingen).
Single-cell suspensions in sodium azide were incubated in the dark for 25 minutes at 37°C with specific antibodies (directly conjugated to fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridin-cholorophyll-protein (PerCP), allophycocyanin (APC), and Pacific Blue, Alexa 700. If a conjugated biotin antibody was used, suspensions were incubated for 25 minutes with streptavidin Qdot800 (Invitrogen), followed by two washes (PBS, 4% sodium azide). The antibodies used were (BD PharMingen or eBiosciences): CD44 (clone IM7), CD4 (clone L3T4), CD8 (clone 53-6.7), CD62L (clone MEL-14), ICAM1 (clone LR16), CD25 (clone 3C7). Intracellular IFNγ was measured by pre-incubating lung cells with monensin (Golgi Stop, BD PharMingen), anti-CD3 and anti-CD28 (4 hours, 37°C, 5% CO2). Cells were surface stained, incubated for 30 minutes (37°C), washed, fixed and permeabilized with Perm Fix/Perm Wash (BD Pharmingen). Finally, the cells were stained with intracellular Foxp3 (clone FJK-16s), IFNγ (clone XMG1.2), TNFα (clone TB 15), IL-4 (clone 16RP) or respective isotype controls (BD Pharmingen) for another 30 minutes. All samples were analyzed on a Becton Dickinson LSR-II and data analyzed using FACSDiva v5.0.1 software. Cells were gated on lymphocytes based on characteristic forward and side scatter profiles. Individual cell populations were identified according to the presence of specific surface markers.
Statistical Analyses
Data are presented using the mean values (n=4) in duplicate or triplicate assays. Chi square test (Gehan-Breslow-Wilcoxon Test) and a nonparametric method using Graphpad Prism 4.0 to determine statistical significance in the survival assay. Significance was determined by calculating P values using an unpaired two-tailed Student t test between groups of data. Values p<0.05 were considered statistically significant.
RESULTS
Increased Susceptibility of Tat-Tg Mice to M. tuberculosis W-Beijing SA161
We first investigated whether there were differences between the Tat-tg and non-tg mice in terms of survival after infection with M.tb W-Beijing SA161. For comparison, mice were also separately infected with H37Rv, a virulent laboratory strain of M.tb. Fig. (1a) shows that while 100% of the non-tg-SA161 infected mice (blue closed circles) survived 65 days post infection, 0% of the Tat-tg-SA161 infected mice (red open squares) were alive at this time (***, p=0.0039; day 65 is indicated by the arrows), suggesting that the presence of Tat and M.tb SA161 infection significantly increases mortality. Non-tg (orange hash marks) and Tat-tg (grey open circles) mice infected with H37Rv, survived infection past the 100-day mark, indicating that increased mortality observed in the SA161 group was associated with the clinical M.tb isolate and the presence of Tat. Because we did not observe a difference in survival between the non-tg and the Tat-tg groups infected with H37Rv, we further examined the consequences of SA161 infection in this murine model, beginning with measuring changes in lung bacterial burden. Although the values for SA161 bacterial load in the lungs were similar for both strains on day 7, there was a trend towards increased bacterial numbers in the Tat-tg mice on days 15 and 30 (Fig. 1b).
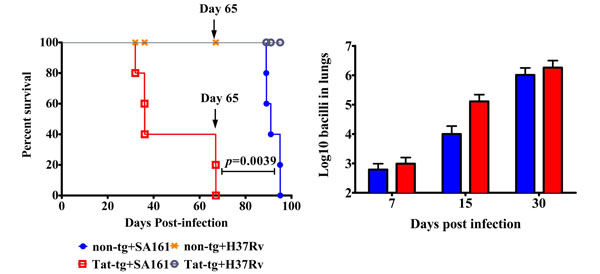
Tat-tg mice infected with M. tuberculosis W-Beijing SA161 show decreased survival compared to non-tg mice, but similar lung bacterial burden. (a) Survival of non-tg and Tat-tg mice was monitored after a low dose aerosol infection with M. tuberculosis W-Beijing SA161 (blue closed circles and red open squares, respectively) or the control laboratory strain H37Rv (orange hash marks and grey open circles, respectively). n=5 mice per group. The arrows indicate day 65 post infection. (b) Bacterial counts from non-tg (blue bars) and Tat-tg mice (red bars) infected with W-Beijing SA161 at the indicated times post infection. Results are expressed as the average Log10 CFUe (±SE) of the bacterial load in each experimental group (n=4).
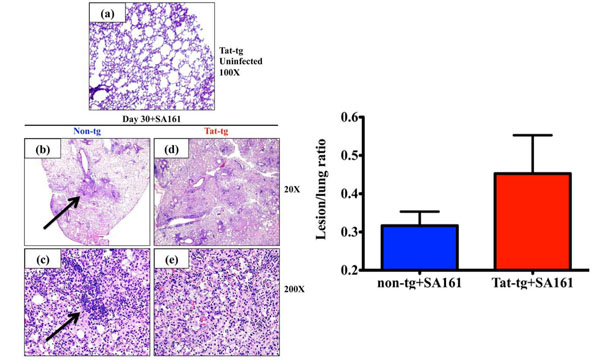
Increased lung pathology in Tat–tg mice infected with M. tuberculosis W-Beijing SA161 compared to non-tg control mice. (a) Representative photomicrograph of H&E-stained lungs from an uninfected Tat-tg mouse. Total magnification a=100x. (b-e) Lung sections from all 4 mice per group infected with a low dose aerosol of W-Beijing SA161 M. tuberculosis 30 days post infection were examined and a representative photomicrographs of H&E-stained lungs from a non-tg (b) and (c) or a Tat-tg mouse (d and e) are shown. Total magnification b, d=20x; c, e=200x. (f) H&E sections were analyzed; total lung area from all 4 mice were measured and compared to areas affected by lesions. The percent lung involvement is reported; SA161-infected Tat-tg mice (red bar), non-tg mice (blue bar).
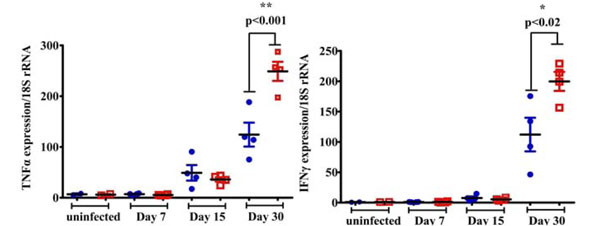
Increased pro-inflammatory cytokine expression in the lungs of Tat-tg mice infected with M. tuberculosis W-Beijing SA161. At various time points post infection (Day 7, 15, and 30), TNFα (a) and IFN-γ (b) mRNA levels were quantified from the lungs of mice. In both cases, the non-tg mice are represented by the blue closed circles and Tat-tg mice are shown as red open squares. (n=4 mice per group). In the absence of bacterial infection, TNFα or IFN-γ expression levels are indicated by the “uninfected” samples shown in both a and b.
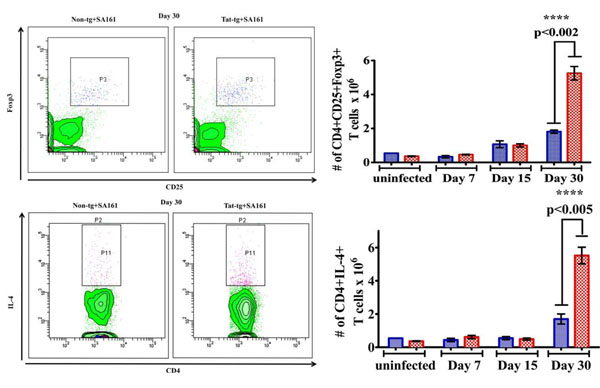
Higher numbers of both CD4+CD25+ T cells that express Foxp3+ and CD4+ T cells that express IL-4 in Tat-tg mice infected with M. tuberculosis W-Beijing SA161. Flow cytometry was used to quantify the number of cells in the lung. CD4+ T cells were first identified by gating on SSC vs CD4+ T cells. >From these, cells that also express CD25+ and Foxp3+ 30 days post infection were gated as shown in the representative dot plot (a). (b) The number of CD4+CD25+Foxp3+ T cells in the lungs of Tat-tg (red open squares) and non-tg (blue closed circles) mice at various time points after infection are reported. Results are expressed as the average number of CD4+CD25+Foxp3 (***, p<0.0002) +/- SEM. (n=4). (c) CD4+ T cells that also express IL-4 were gated; a representative blot at day 30 is shown. (d) The number of CD4+IL-4+ T cells in the lungs of non-tg and Tat-tg mice after infection were quantified at various time points. Results are expressed as the average number of CD4+IL-4 (***, p<0.0005) cells in each group, +/- SEM. (n=4).
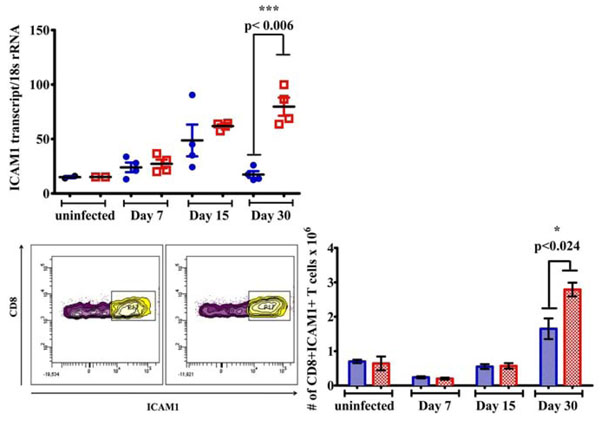
Increased ICAM1 expression in Tat-tg mice infected with M. tuberculosis W-Beijing SA161. (a) Lung expression of ICAM1 by SA161-infected Tat-tg (red open squares) and non-tg (blue closed circles) mice was measured by qRT-PCR (**, p =0.006). Samples labeled “uninfected” represent mice that were not infected with mycobacteria. (b) ICAM1+ CD8+ T cells were also gated; a representative blot at day 30 is shown. (c) The total number of ICAM1+ CD8+ T cells in the lungs of Tat-tg (red) and non-tg mice (blue) after infection are shown (n=4). Results are expressed as the average number of ICAM1+ CD8+ T cells, +/- SEM. Samples labeled “uninfected” represent mice that were not infected with mycobacteria.
Increased Lung Pathology in Tat-Tg Mice Infected with M. tuberculosis W-Beijing SA161
Histological examination of lungs from uninfected Tat-tg mice shows normal tissue architecture and the absence of notable inflammatory infiltrates or granulomata (Fig. 2a). To determine if the accelerated mortality of the Tat-tg mice was associated with histopathologic changes, lung tissues were collected 30 days post infection and stained with H&E. Consequently, all sections were examined and representative sections are shown. While the non-tg lung (Fig. 2b) show organized granulomata-like lesions as indicated by the arrows and multiple areas of uninvolved lung space, the lung lesions of the Tat-tg mice were extensive, more diffuse and disorganized, with few, if any, organized granulomata-like lesions (Fig. 2d). When the sections were observed under a higher magnification, the non-tg lungs show organized lesions characterized by the presence of coalescing inflammatory foci and lymphocytic aggregates (Fig. 2c, arrow). On the other hand, Tat-tg mice show disorganized lesions and a lack of lymphocytic aggregation (Fig. 2e). To gain a better understanding of the impact infection had on the lung tissue at day 30, we determined the fraction of lung parenchyma affected by granulomatous inflammation in both mouse strains. At 30 days post infection the mean percent involvement of the lung by inflammation in the non-tg group was 31% compared to 45% for the Tat-tg mice (Fig. 2f), but this difference was not statistically significant.
Higher Levels of TH1 Cytokines and Elevated TH2 Immune Responses in the Lungs of Tat-tg M. tuberculosis W-Beijing SA161 Infected Mice
The balance between TH1 and TH2 immune responses influences the coordinated and dynamic events involved in granulomata development. The production of tumor necrosis factor α (TNFα), IL-12, and interferon-gamma (IFNγ) cytokines associated with the TH1 response is crucial in controlling M.tb [32]. Such pro-inflammatory responses involve the recruitment of antigen-specific T lymphocytes and interactions with macrophages and dendritic cells [33, 34]. TH2 responses that follow include T regulatory cells, IL-10 and IL-4 cytokines [35] that downregulate TH1-dependent responses [30, 36]. We used qRT-PCR, flow cytometry, and intracellular cytokine staining to dissect the phenotypes of immune cells in the lungs during the course of infection.
We found that expression levels of the pro-inflammatory cytokines TNFα and IFNγ in the lungs did not significantly differ between the non-tg and Tat-tg groups in the absence of bacterial infection (Fig. 3a, b, “uninfected”). In contrast, we observed higher TNFα (Fig. 3a**, p=0.001) and IFNγ (Fig. 3b, *, p=0.02) mRNA expression in the Tat-tg mice infected with M.tb W-Beijing SA161, compared to the non-tg/infected mice 30 days post infection. Although the qRT-PCR results do not allow for discrimination of the cellular populations responsible for cytokine production, it does suggest that the presence of Tat alters the pro-inflammatory cytokine response during M.tb infection. Unfortunately, all available lung tissues were consumed in the assays reported here and we were unable to measure TNFα and IFNγ protein levels.
To better understand the role inflammatory cells play in the lung after infection with M.tb W-Beijing SA161, we quantified the numbers of TNFα, IL-12, or IL-10+ producing CD11c-CD11b+ macrophages, CD11c+CD11b-DEC205+ dendritic cells, and CD4+ and CD8+ T cells that express CD44hiCD62Lhi or CD44hiCD62Llo (representing memory and effector T cells, respectively). In general, we found no significant differences between the non-tg and Tat-tg mice in the relative number of these cell types in this tissue over time (data not shown).
T regulatory cells control over-stimulated immune responses by maintaining the physiologic equilibrium between the immune system and cell homoeostasis [37]. In particular, CD4+CD25+ T regulatory cells have roles in the immune response to M.tb [30] and are important producers of immunosuppressive cytokines (such as IL-4). Foxp3-expressing CD4+CD25+ T cells are widely accepted as the definitive marker for T regulatory cells [37], but their role in tuberculosis and HIV infection remains poorly defined. Thus, we monitored the kinetics of these cells post infection using flow cytometry. SSClow lymphocytes vs CD4+ T cells in the lung were identified; from this population, cells that were also CD25+Foxp3+ (Fig. 4a) were gated (representative plots from day 30 are shown). We found Tat alone did not impact the number of CD4+CD25+ Foxp3+ T regulatory cells (Fig. 4b, “uninfected”), but significantly greater number of these cells were found in the Tat-tg mice 30 days post infection compared to non-tg mice (***, p<0.002). Similarly, CD4+ T cells that were also IL-4+ were gated (Fig. 4c). Comparable numbers of these cells were found in the absence of bacterial infection (Fig. 4d, “uninfected”), but were significantly higher in the Tat-tg mice 30 days post infection (Fig. 4d, ***, p<0.005), suggesting the presence of Tat and SA161 results in higher number of suppressive cells, which may contribute to increased mortality.
Increased ICAM1 Expression in Tat-Tg M. tuberculosis W-Beijing SA161 Infected Mice
We next investigated a possible explanation for the increased numbers of CD4+CD25+ Foxp3+ T regulatory and CD4+IL-4+ producing T cells in the lungs of the Tat-tg mice. In a previous report, Windish et al. indicated ICAM1-/- mice show fewer numbers of CD4+CD25+ Foxp3+ T cells when compared to wildtype controls after infection with M.tb, suggesting ICAM1 may be involved in the recruitment of these cells to the lung upon exposure to the pathogen [39]. Employing quantitative PCR, we found significantly higher ICAM1 lung expression in the M.tb W-Beijing SA161 infected Tat-tg mice compared to the non-tg mice 30 days post infection (Fig. 5a, **, p<0.006); the presence of Tat alone in lung did not alter ICAM1 expression (Fig. 5a, “uninfected”). Because CD4+, CD8+ T cells are important in the adaptive immune response to M.tb, the numbers of CD4+ and CD8+ cells expressing ICAM1 were also monitored. Although we found no difference in CD4+ICAM1+ T cell numbers between the mouse strains (data not shown), increased numbers of lung CD8+ICAM1+ T cells were observed in the Tat-tg mice compared to non-tg animals 30 post-infection (Fig. 5c, * p<0.024) when CD8+ICAM1+ T cells were gated (Fig. 5b).
DISCUSSION
Tuberculosis has emerged over the past decade as the most prominent bacterial disease found in HIV-positive individuals worldwide [40, 41]. A relevant animal model of these co-infections would be highly useful; but whereas M.tb infects mice, HIV does not. An alternative approach is to derive a transgenic mouse that possesses organ-specific expression of key HIV proteins. Tat is a prime viral protein candidate because it is secreted from infected cells and taken up by uninfected bystander cells. Pulmonary complications cause morbidity and mortality in HIV-infected individuals even in the HAART era [42]. These complications are not only attributable to the direct presence of HIV in the lungs, but secreted viral proteins such as Tat. In the murine model used here, Tat is expressed in the lungs, providing a useful tool to study host immune responses to aerosol bacterial infection and to monitor its in vivo effects. Although it is difficult to accurately quantify the amount of Tat in the lung tissue of the Tat-tg mice, we have previously detected immunoreactive Tat protein from lung tissue via immunoblotting [28]. Using densitometry, we postulate the protein level of Tat in these mice is approximately 1-5ng/mg of total lung protein (unpublished). Westendorp et al. previously reported the circulating level of Tat from AIDS patients to be approximately 1.6ng/ml [43]. It is difficult to discern whether infection with W-Beijing SA161 impacts Tat expression or the levels of endogenous Tat protein; thus it would be sensible to monitor changes in the amounts of this viral protein in future studies. At present, we hypothesize Tat contributes to histopathological changes in the lungs and hampers efficient immune responses to M.tb.
Infection of animal models with laboratory strains of M.tb such as H37Rv may not adequately model the pathogenesis of naturally occurring tuberculosis isolates; thus we evaluated the immune response to M.tb W-Beijing SA161 infection. However, as a comparison, groups of non-tg and Tat-tg mice were also infected with H37Rv and their survival monitored. The significant finding from this study was that the Tat-tg mice were more susceptible to infection with W-Beijing SA161, as the mean survival time of Tat-tg mice was approximately 30 days, a considerably shorter survival time than the non-tg mice (Fig. 1a). Because necropsies were not completed, we do not know the exact cause of death, but it is possible lung consolidation is a major contributing factor. Also, because the lung was the only tissue in which bacterial load was monitored, the extent of extra-pulmonary dissemination of the bacteria in this experiment is not known. In contrast, all mice infected with H37Rv lived past the 100-day mark, suggesting infection with W-Beijing SA161 contributed to the quickened time to death in both groups of mice. A limitation to our survival assay was the small number of mice used per group, however we believe five mice per group was sufficient because a significant difference in survival between the Tat-tg and non-tg mice after W-Beijing infection (****, p=0.0039) was observed. These data are consistent with previously published work [44-46] that similarly reported increased mortality of mice infected with W-Beijing strains. Despite the differences in survival, the lung bacterial load at day 30 did not significantly differ between the two groups (Fig. 1b), however, the bacterial burden increased steadily up to this time point. These findings are consistent with previous reports of the rate of growth of other laboratory M.tb isolates after aerosol infection of non-tg mice [47]. These results suggest that bacterial burden is not the principal factor for mortality, but rather, we propose that Tat dysregulates the immune response and impairs immunity to bacterial challenge.
While histopathological analysis demonstrated that the uninfected Tat-tg mice show normal lung space with healthy alveoli (Fig. 2a), we did observe gross differences in the architecture of the lung granulomata-like lesions between the W-Beijing SA161-infected non-tg and Tat-tg mice (Fig. 2b-e). Non-tg lungs showed moderate, discrete foci of inflammatory infiltrates, suggestive of granulomata-like lesions. These findings are consistent with previous reports from co-author Ordway et al. regarding the pathology of C57BL/6 mice infected with a “hypervirulent” M.tb W-Beijing isolate, HN878, at this timepoint [30]. Per Ordway et al. macrophages and neutrophils likely comprise the inner most portion of the granuloma, surrounded by a “perivascular cuff” of newly expanded antigen-specific T cells [30]. Although it is difficult to do the side-by-side comparisons of our findings to Ordway et al., we believe Tat excessively recruits inflammatory and/or immunosuppressive cells to the site of infection, consequently resulting in compromised lung function. The Tat-tg granulomata were significantly less well-organized and showed considerable areas of diffuse inflammatory infiltrates, suggestive of lung consolidation, with very few sections of normal tissue. When the percentage of lung lesions at day 30 in each group was calculated, increased lung involvement in the Tat-tg group was confirmed (Fig. 2f). It is likely the bacteria disseminated to extra-pulmonary sites in the Tat-tg mice, however, since we did not culture the bacteria from locations other than the lungs, it is difficult to determine whether containment of the bacteria to the lungs was compromised; nonetheless dissemination is probable.
qRT-PCR was used to examine the changes in gene expression after M.tb W-Beijing SA161 infection. Analysis revealed increased expression of TNFα and IFNγ in the infected Tat-tg mice (Fig. 3), indicating that the combination of Tat and bacterial infection, but not Tat alone, alters the cytokine milieu. Although we did not quantify the protein levels of these pro-inflammatory cytokines, Ordway et al. confirmed that C57BL/6 mice infected with HN878 demonstrated significant production of IFNγ and TNFα early after infection [30]. TNFα and IFNγ responses have been shown to be important in host defense against M.tb [48, 49]. Although we observed increased expression of TNFα and IFNγ in the M.tb W-Beijing SA161-infected Tat-tg mice as compared to the non-tg group, we also noted a paradoxical decrease in survival. The reason for this is unknown, however, it is reasonable that infection with W-Beijing M.tb compromises lung function and architecture, concomitant to the onset of decline in pro-inflammatory responses.
Flow cytometry was used to elucidate the cellular populations after bacterial infection. While we did not observe significant differences in the number of macrophages, dendritic cells, or activated T cell populations in the lungs, we did find dramatically higher numbers of immunosuppressive cellular phenotypes including CD4+ CD25+Foxp3+ and IL-4 producing CD4+T cells in the Tat-tg mice 30 days post infection (Fig. 4b, d, respectively). Of note, the presence of Tat alone did not alter the cellular milieu in the absence of SA161 infection (Fig. 4b, d, “uninfected”). Our findings are again consistent to that of Ordway et al. who also observed a similar increase in the numbers of lung CD4+CD25+Foxp3+ T regulatory cells, dampened survival, and severe lung pathology in HN878-infected mice, despite a robust TH1 response [29]. Our data do not allow us to discriminate where these infiltrating cells originate from, nor do they definitively prove increased numbers of CD4+CD25+Foxp3 T cells cause lung injury. It is possible that the heightened numbers of CD4+CD25+ Foxp3+ cells compromise lung function due, in part, to Tat-mediated increase of adhesion molecules that influence the adhesion of infiltrating cells in the lung such as ICAM1-expressing cells (Fig. 5a), specifically ICAM1+ CD8+ T cells (Fig. 5c). Liu et al. reported that Tat induces VCAM1 expression in human pulmonary artery endothelial cells [50] that facilitates lymphocyte attachment in the lung. D’Alessio et al. established an association between the presence of CD4+CD25+Foxp3 T cells and lung damage using a well-established model of acute lung injury via exposure to lipopolysaccharide (LPS) [51]. Moreover, this group detected the presence of T regulatory cells in the alveolar compartment of humans with acute lung injury.
Herein, we characterize the susceptibility and early immune responses (up to 30 days post infection) of non-tg and Tat-tg mice to low-dose aerosol of M.tb W-Beijing SA161. In parallel, we also examined the immune response to M.tb H37Rv; however we did not note any significant differences between the Tat-tg and non-tg mice infected with H37Rv in any of the parameters tested and were not pursued further. We show that M.tb W-Beijing SA161-infected non-tg mice form well-structured granulomata due to the organized and coordinated balance of the pro-inflammatory and immunosuppressive immune responses. This balanced immune response enables the non-tg mice to survive longer than the Tat-tg mice. Conversely, the Tat-tg mice succumbed to infection sooner, but this was not due to increased lung bacilli, since lung CFU counts were similar between the non-tg and Tat-tg mice. Rather, we posit that the presence of Tat in the lungs leads to increased ICAM1 expression resulting in the excessive recruitment of Foxp3 T regulatory and CD4+ producing IL-4+ cells to the lung. Influx of these cells into the lung results in disorganized and diffuse lesions that likely compromise lung function. While in the lung, these suppressive cells produce mediators that likely dampen pro-inflammatory responses at a time when these defenses are leveling off or decreasing. We postulate the aforementioned is mediated by TGF-β. In fact, Song et al. has documented that Tat enhances ICAM1 expression in astrocytes [25] and Windish et al. has reported ICAM1 is required for the induction of Foxp3 T regulatory cells through mechanisms mediated by TGF-β [39]. Taken together, these factors contribute to homeostatic imbalance, compromised lung function, and accelerated mortality. Although we did not directly assay for TGF-β in our model, Tat has been shown to induce TGF-β [52]. It is possible this disproportionate number of Foxp3 T regulatory cells and CD4+ IL-4 producing T cells likely suppresses ongoing TH1 responses at a critical time at which these responses are abating, resulting in containment failure. In fact, a report by Sharma et al., demonstrated recovery of CD4+CD25+Foxp3+ T regulatory cells from bronchoalveolar lavage fluid from miliary tuberculosis patients [53].
At this time, we do not know the events that transpire in the W-Beijing SA161-infected mice after 30 days of infection. We anticipate that disease in the non-tg mice will progress in a manner consistent with previously published reports of murine tuberculosis using other M.tb W-Beijing strains. Previously, Ordway et al. demonstrated that at 100 days post infection, C57BL/6 mice infected with M.tb W-Beijing HN878, show larger and more numerous lung lesions that resulted in complete lung consolidation [29]. In terms of the immune response, Ordway et al. reported lung cells isolated from C57BL/6 mice infected with M.tb HN878 or H37Rv show comparable levels of IFNγ, TNFα, MCP-1, and IL-6 at timepoints taken later than 30 days post infection, but heightened T regulatory cell responses with HN878 infection. We expect that if we had performed longer-term studies, similar immunological responses would have developed in the M.tb W-Beijing SA161-infected non-tg mice used in our study at these later timepoints.
Based on our findings, we believe the presence of Tat in the lung dysregulates the normal pro-inflammatory and suppressive immune responses generated in response to infection with M.tb W-Beijing SA161.
CONCLUSION
The loss of CD4+ T cell responses in HIV-positive individuals is the basis of the increased susceptibility to multiple opportunistic infections. It is becoming increasingly evident that a range of end-organ pathologies, including the lung, cannot be directly ascribed to the presence of the virion at the tissue site. The new approach described here, using a mouse that expresses a key viral protein in the lungs, may provide new information about the pathogenesis of co-infections. We have been able to demonstrate that the presence of Tat disrupts the normal host immune response to M.tb. To our knowledge we are the first to report a link between HIV-Tat and CD4+CD25+Foxp3+ T regulatory cells and increased lung pathology. It will be prudent to confirm the role of ICAM1 in the recruitment of Foxp3 T regulatory cells using neutralizing antibodies or siRNA against ICAM1 in future studies. In addition, future work should define the molecular mechanisms that govern the association between Tat and this particular population of adaptive T cells. Taken together, these data suggest that Tat influences and disrupts the orderly emergence of acquired immunity to tuberculosis infection.
ACKNOWLEDGEMENTS
This study was funded by the NIH National Heart Lung and Blood Institute #5R01HL059785. The authors would like to thank Dr. Adela Cota-Gomez for helpful discussions and critical review of the manuscript and Melanie Hakar for her assistance with histopathology.
Jennifer R. Honda, Ph.D. is supported by an NIH NRSA Infectious Disease Training Award, T32-AI007447-19.

