All published articles of this journal are available on ScienceDirect.

A Multicenter, Open Labeled, Randomized, Phase III Study Comparing Lopinavir/Ritonavir Plus Atazanavir to Lopinavir/Ritonavir Plus Zidovudine and Lamivudine in Naive HIV-1-Infected Patients: 48-Week Analysis of the LORAN Trial
Authors Info & Affiliations
Abstract
Objective:
The primary aim of the study was to compare the metabolic side effects of a nucleoside analogue-containing regimen with a nucleoside analogue-sparing double protease inhibitor regimen. A secondary goal was to test for efficacy of a double-PI regimen.
Design:
Multicenter, randomized, open-label, phase III clinical trial.
Subjects:
Adult HIV-1-infected individuals naïve to antiretroviral therapy with viral load above 400 HIV-RNA copies/ml were randomized (1:1) to either 400 mg lopinavir /100 mg ritonavir (LPV/r) BID plus 150 mg lamivudine/300 mg zidovudine (CBV) BID versus LPV/r BID plus 300 mg atazanavir (ATV) QD. Main outcome measure was the virologic failure in both groups, defined as viral load ≥50 copies/ml at week 48.
Results:
In the CBV/LPV/r-arm, 29 out of 35 patients [(83%; 95% confidence interval (CI) 66.9-92.2%] and 18 out of 40 patients (45%; 95% CI 29.7-61.5%) in the ATV/LPV/r-arm had a HIV-RNA level <50 copies/ml at week 48. The intent-to-treat analysis revealed inferior virologic response in the ATV/LPV/r arm (Chi-Q and Fisher´s Exact Test p<0.001) and resulted in premature termination of the trial. Eleven patients in the ATV/LPV/r-arm discontinued therapy because of virological failure. These failures mostly presented with low level replication (<1,000 copies/ml). Increases in CD4 cell counts was significantly more rapid in the ATV/LPV/r arm (p=0.02), but comparable at week 48.
Conclusions:
ATV/LPV/r had less virologic efficacy than the conventional RTI-based regimen and resulted in a high virological failure rate with low level replication.
INTRODUCTION
Highly active antiretroviral therapy (HAART) has been proven very efficacious for long-term control of viral replication and disease progression in HIV-infected individuals. The International AIDS Society currently recommends standard combination regimens including two nucleoside reverse transcriptase inhibitors (NRTI) plus either one protease inhibitor (PI) or one non-nucleoside reverse transcriptase inhibitors (NNRTI) in therapy-naïve patients [1]. Numerous studies provide evidence for the excellent antiviral activity of these combinations and support the preferred application in first-line therapy of HIV-infected patients. However, conditions, such as adverse side effects and virologic failure leading to resistance development, constitute major confinements in HIV treatment, thereby limiting therapeutic options in a significant number of patients. Particularly, peripheral lipoatrophy, hepatic steatosis and metabolic disturbances including hyperlipidemia, insulin resistance, and lactic acidosis have been attributed to both NRTI-and PI-containing regimens [2-5]. The pathogenesis of these adverse effects during antiretroviral therapy has not been fully elucidated yet, but mitochondrial toxicity due to thymidine analogues has been proposed and replacement of these compounds by alternative NRTIs has been demonstrated to result in partial recovery of lipoatrophy and lipid disturbances [4-6] and NRTI-sparing regimens are now being explored in clinical trials.
The other major obstacle is the development of viral resistance leading to early virologic failure of HIV-therapy. This may be due to insufficient adherence, but also virally inherited causes or pre-existent mutations may play a role [7]. In this regard, resistance due to escape mutations in the HIV reverse transcriptase gene is a common phenomenon of failing antiretroviral therapy in patients receiving NRTI-containing HAART. Several studies support the notion that regimens containing boosted PIs postpone the incidence of NRTI resistance for many years or during the early phases of viral rebound.
Based on these observations and more recent data [7], certain triple NRTI therapies appear to have inferior efficacy [1].
The aim of this study was to compare the effects of a NRTI-sparing regimen [lopinavir/ritonavir (LPV/r) plus atazanavir (ATV)] versus a NRTI-containing regimen (LPV/r plus lamivudine and zidovudine) in regard to efficacy, toxicity, and metabolic parameters in ARV-naïve patients. This national multicenter randomized study addresses the following question: Do metabolic parameters, clinical side effects and quality of life (QoL) differ between a NRTI-containing regimen and a NRTI-sparing regimen? Since we assumed the beginning of the study no problems with virological efficacy of the regimens, virological response was a secondary goal only.
METHODS
Subjects and Design
The LORAN study was a multicentre randomized, open-label study in which co-administration of LPV/r and ATV (ATV/LPV/r) was compared to LPV/r combined with fixed-dose combination of zidovudine plus lamivudine in HIV-positive, antiretroviral-naïve patients. Subjects were randomized (1:1) to receive either 400 mg LPV/100 mg ritonavir BID (Kaletra®) plus 150 mg lamivudine/300 mg zidovudine BID (Combivir®, CBV) versus 400 mg LPV/100 mg ritonavir (Norvir®) BID plus 300 mg ATV (Reyataz®) QD. A total of 75 subjects were studied for a study period of 48 weeks.
Inclusion criteria were: HIV-patients at age ≥18 years, naive to antiretroviral therapy, not pregnant or breastfeeding, and not having been treated for an active AIDS-defining opportunistic infection or receiving any investigational drug within 30 days of screening. HIV-RNA level had to be above 400 copies/ml. Exclusion criteria were severe changes in blood count, liver or renal impairment, cardiomyopathy (due to any cause) or any significant cardiovascular or electrophysiological disease. Baseline data, including demographic data, physical and laboratory status, and status of disease according to 1993 Centres for Disease Control and Prevention classification, were obtained before initiation of HAART. All subjects, regardless of treatment assignment, returned to the site for a week 4 and week 12 study visit and then every 12 weeks until week 48. Procedures at these visits included physical examination, vital sign measurements, clinical laboratory tests and determinations of antiviral and immunologic activity. The study protocol was approved by the ethics committee of the Hannover Medical School and by the local ethics committees of the investigational sites. The study was conducted in accordance with the protocol, ICH GCP, regulations governing clinical study conduct, ethical principles that have their origin in the Declaration of Helsinki, currently 1996 revision and all applicable local regulations. All patients gave written informed consent before recruitment. The primary endpoints were determined as the differences of the NRTI-containing or dual PI regimen on metabolic parameters (glucose metabolism, lipid paramenters), body fat compositions and quality of life. The planned sample size of at least 50 subjects in each treatment group provided a 80% power (β=0.80) to detect a difference in glucose and lipid levels based on the study published by Saint-Marc et al. [8]. Assuming metric scale of the relevant parameters, normal distribution and similarity of variance in both study arms, 50 subjects in each study arm should be enough to provide a 80% power to detect differences of 20-30% for other clinically relevant parameters based on an a priori analysis performed with Clinical MEDORA Software.
Randomization was performed by means of a predefined randomization list (1:1 ratio), using a central randomization system (facsimile response) to ensure balance across treatment groups in each stratum. Combivir® and Kaletra® were prescribed by the investigational sites, Reyataz® was bought wholesale and was provided by the Principle Investigator. At each study visit, patients underwent routine safety monitoring, including hematology and chemistry evaluations, CD4 T-cell count, and plasma HIV-RNA levels. Measurements were performed at local laboratories. Samples for pharmacokinetic measurements were alternating drawn, centrifuged, frozen and sent to for further analysis to a central lab. Drug monitoring was not regularly performed and provided in cases of treatment failure.
Monitoring and data collection at the investigational sites was performed by Abbott co-workers. An independent Data and Safety Monitoring Board had access to the data, reviewed the data and performed the statistical analysis at the planned interim analysis.
Study End Points
The primary endpoints were determined as the influence of NRTI on metabolic parameters and body fat compositions and quality of life. The secondary endpoints were the clinical side effects, viral suppression at week 48, and immune reconstitution at week 48. Virologic failure was defined using a sensitive test system (limit of detection <50 HIV-RNA copies/ml) as either: a) a failure to reach a viral load below the detection limit by week 48, or b) a confirmed reincrease of HIV-RNA from the not measurable to the measurable range.
A topical interim analyses proved statistical significance for the virologic failure in both therapy arms at week 24 with a higher rate in the ATV/LPV/r arm than in the CBV/LPV/r arm. The statistical calculations were provided by a member of the Data Safety Monitoring Bboard (DSMB). The statistical difference in the ITT analysis showed p=0.021, the observed failures p <0.001, in each case calculated using the Fisher's Exact test. Therefore recruitment for this prospective, randomized trial was terminated in November 2006.
This of course had enormous consequences for the study. The original primary goals could not be reached and therefore the virological efficacy became the primary focus of our study.
Resistance Determination
For resistance determination, viral genotypic assessment was performed by Fenner Laboratory, Hamburg, Germany, and by the Institute for Virology, University of Cologne, Germany, using different algorithms (Geno2Pheno, ANRS, Stanford Database).
Plasma viral load was measured with a commercial kit in use in each participating centre or in a cooperating laboratory; all had a level of detectability of 50 copies/ml. In patients experiencing virologic failure, HIV-1 resistance genotyping was performed on frozen plasma samples obtained at the time of HIV-RNA increase, in parallel with the baseline sample.
Pharmacokinetic measuring was performed by a single cooperating laboratory using LC-MS/MS technique. Samples for pharmacokinetic measurements were drawn at weeks 4, 24, 28, and 48 before and three hours after intake of study drugs. Sera were frozen at minus 20 °C, collected and sent to the principal investigator’s laboratory. In seven subjects in the double-PI failures, lopinavir and atazanavir levels were measured.
Statistical Analysis
The primary endpoint for which this study was powered could not be reached since the study had to be stopped prematurely. Therefore the originally secondary endpoint, the virological efficacy, became the primary variable to be analyzed. Therefore we analyzed the proportion of subjects with a plasma HIV-RNA level below 50 copies/ml at week 48. An intent-to-treat approach was used for this analysis in which missing values were considered above 50 copies/ml unless the immediately preceding and immediately following values are below 50 copies/ml (“non-completer=failure” analysis). All values obtained more than one day after study drug discontinuation were considered above 50 copies/ml. The estimate of the proportion of subjects with HIV-RNA levels below 50 copies/ml is provided for each treatment arm, along with the corresponding 95% confidence interval for the difference in proportions based on the normal approximation to the binomial distribution. The proportion of patients with HIV-RNA below 50 copies/ml at each visit was summarized using an intent-to-treat (missing=failure) method, in which all missing values were considered above 50 copies/ml, an intent-to-treat (last observation carried forward) method, in which missing values was replaced by the most recent non-missing value, and an observed data method, in which missing values were excluded from the analysis. Comparisons between treatment arms were performed using Fisher’s exact test. Mean changes from baseline to each protocol-specified visit were analyzed using a one-way analysis of variance (ANOVA) model with treatment arm as the only factor. Analyses included all data obtained while subjects were on randomized antiretroviral therapy. The level of significance was set at p<0.05. Statistical analysis was performed using SPSS 16 (SPSS Inc., Illinois).
RESULTS
Study Population
From September 2004 until November 2006, 80 consecutive HIV-infected HAART-naïve patients at 14 sites fulfilled inclusion criteria and were recruited and assigned randomly to either treatment arm. Two patients each revoked consent prior to and immediately after baseline visit, respectively, one was lost to follow up immediately after baseline. Seventy-five patients were enrolled in the study and began intake of study drugs. The baseline characteristics of these 75 patients are provided in Table 1. Patients were mainly male (13.3 % female), and their mean age was 39 years. At baseline, the mean plasma HIV-1 RNA level was log 5.3 copies/ml, and the mean CD4 cell count was 220 cells/µL. Baseline characteristics were well balanced among the two study groups. A total of 29 (83 %) in the CBV/LPV/r -arm and 21 (53 %) in the ATV/LPV/r -arm completed the protocol until week 48.
Baseline Characteristics of Study Population
| Baseline Characteristics | All Patients | CBV/LPV/r Group | ATV/LPV/r Group |
|---|---|---|---|
|
|
|||
| Total number | 75 | 35 | 40 |
|
|
|||
| Female – no. (%) | 10 (13.3) | 5 (14.3) | 5 (12.5) |
|
|
|||
| Age (years) (Mean (range)) | 39.1 | 36.6 (25-65) | 41.4 (27-66) |
|
|
|||
| CD4 count | |||
| Mean | 220 | 239 | 203 |
| 0-99 cells/mm3 | 12 | 5 | 7 |
| 100-199 cells/mm3 | 20 | 7 | 13 |
| 200-399 cells/mm3 | 40 | 21 | 19 |
| ≥400 cells/mm3 | 3 | 2 | 1 |
|
|
|||
| HIV-1 RNA level | 208,463 | 142,422 | 270,836 |
| (Mean (range)) | (745-4,200,000) | (2,623-548,000) | (745-4,200,000) |
| log | 5,319 | 5,154 | 5,433 |
| <50,000 cop/ml | 27 | 13 | 14 |
| 50,000-99,999 cop/ml | 12 | 6 | 6 |
| 100,000-299,999 cop/ml | 26 | 12 | 14 |
| ≥300,000 cop/ml | 10 | 4 | 6 |
|
|
|||
| Clinical category | |||
| A | 45 | 22 | 23 |
| B | 24 | 10 | 14 |
| C | 6 | 3 | 3 |
Treatment Limiting Events and New Grade 3 or 4 Clinical or Laboratory Abnormalities
1 p=0.0008;
2 including, airway infection, sepsis, perineal abscess.
Virologic Response
For primary efficiency intent-to treat analysis at week 48, 29/35 [80%; 95% confidence interval (CI) 66.9-92.2%] of patients had a HIV-RNA level below 50 copies/ml in the CBV/LPV/r-arm and 18/40 (45%; 95% CI 29.7-61.5%) in the ATV/LPV/r-arm, respectively (Fig. 1). This difference was statistically significant according to chi-square test and to Fisher´s exact test (both p<0.001). When the definition for virological failure also considered patients with a detectable viral load after previous viral suppression below 50 copies/ml, 24/35 (69%) of patients receiving CBV/LPV/r had a viral load below 50 copies HIV-RNA/mL at week 48, as compared to 16/40 (40%) in the ATV/LPV/r-arm. When all patients still receiving study medication at week 48 were considered (observed data analysis), 29/29 (100%) of patients in the CBV/LPV/r-arm had a viral load below 50 copies/mL as compared to only 18/21 (86%) in the ATV/LPV/r-arm.
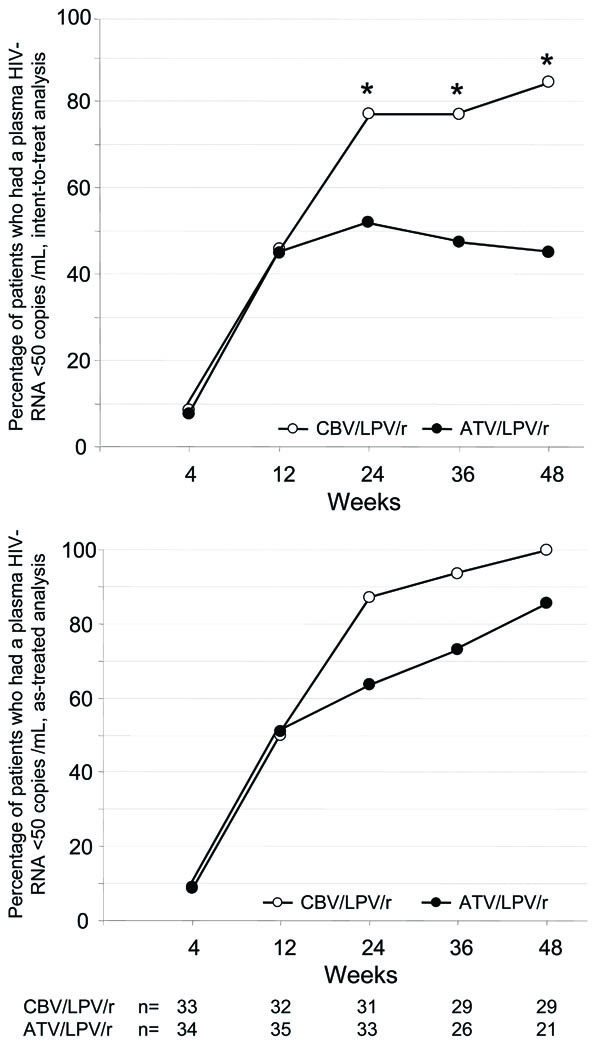
Virologic response. Virologic response as percentage of all HIV-1-infected patients with a viral load below 50 copies HIV-RNA/mL plasma after enrolment into LORAN study (“intend-to-treat” analysis, top panel) and in all patients receiving antiretroviral therapy in both arms (“observed data” analysis, bottom panel).

Immunologic response. Time course of mean (SEM) CD4 cell counts (top panel) and mean (SEM) change in CD4 cell counts from baseline (buttom panel) in both treatment arms.
There was a trend (p=0.077) for delayed time to viral suppression in the double-PI-arm. At week 24, 12 of 33 patients (36%), which were still on study medication, continued to have a detectable viral load in the ATV/LPV/r arm (range 71-3,420 copies HIV-RAN) as compared to 4 of 31 patients (13%) receiving drugs in the CBV/LPV/r arm (range 53-90 copies HIV-RNA). Of the 12 patients with detectable viremia in the ATV/LPV/r arm, 11 had a viral load between 50 and 1,000 HIV-RNA copies/ml, but all patients with detectable viremia in the CBV/LPV/r arm had less than 100 copies HIV-RNA/ml. In addition, 8/33 patients (24%) receiving ATV/LPV/r at week 24 had an increasing viral load as compared to only 1/31 of patients (3%) treated with CBV/LPV/r. Therefore the drug safety monitoring board (DSMB) recommended termination of patient recruitment. Overall 11 patients in the ATV/LPV/r-arm discontinued therapy before week 48 because of virological failure, but none of the patients in the CBV/LPV/r-arm.
Subsequently nine out of 33 patients still on ATV/LPV/r therapy at week 24 were consecutively switched to an alternative regimen, but were not followed up until week 48. Seven of these nine patients had a rebound in viral load before week 36 and before switch. Two out of three patients with a detectable viral load in the ATV/LPV/r arm at week 48 had shown a viral rebound from below 50 copies/ml at week 24. Altogether, eleven patients were discontinued due to virologic failure, all of them in the ATV/LPV/r arm. In agreement with the Drug Safety Monitoring Board, all subjects in the ATV/LPV/r arm were offered the possibility to switch to CBV/LPV/r independent of virologic success.
Immunological Response
With respect to the immunological response, the increase of the CD4+ T cell counts was more rapid up to week 48 in the ATV/LPV/r arm (p=0.02) in the “observed data” analysis (Fig. 2). At week 48, however, the mean increase from baseline was comparable between groups with 151 cells per cubic millimetre (standard error of the mean (SEM)) 125.1, range -74 – 412) in the ATV/LPV/r group, and 142 (SEM 99.4, range -171 - 269) in the CBV/LPV/r group, respectively (Fig. 2).
Pharmacology and Genotypic Resistance
In three out of seven patients with virologic failure amplification of HIV was successful. No major genotypic changes in HIV-1 protease gene were detected at failure in these three patients. Merely, one NRTI resistance (M184IM) was detected in a patient receiving ATV/LPV/r. Pharmacokinetic measuring showed slightly reduced LPV concentrations (2,900 ng/ml) in 1/7 tested subjects in the double-PI failures. This patient confirmed wanting adherence in the intake of study drugs and had normal lopinavir level (4,360 ng/ml) four months later.
Safety Results
In 13 of the 75 patients (17.3 %) toxicity led to discontinuation of one or more study drugs occurred; 7/40 (17.5 %) in the ATV/LPV/r arm and 6/35 (17.1 %) in the CBV/LPV/r arm, respectively. Two individuals stopped therapy because of diarrhea in the CBV/LPV/r group versus one in the ATV/LPV/r group. Three patients in the CBV/LPV/r arm suffered from severe nausea and one developed anemia. In the ATV/LPV/r group, four patients discontinued due to hyperbilirubinemia, one had rash, and one was discharged because of hypertriglyceridemia (Table 2).
A total of 8 of 75 patients (10.3 %) presented with new grade 3 or 4 clinical or laboratory abnormalities leading to hospitalization, but not to termination of study medication. In the CBV/LPV/r arm, two patients suffered from gastrointestinal discomfort, one developed anemia during co-administration of ribavirin for hepatitis C therapy and another one was admitted to hospital for an airway infection. In the ATV/LPV/r arm, one subject became conspicuous with true sepsis due to a dental focus and another one with perineal abscess, which was already evident prior to commencement of study drugs. All these patients recovered totally. An association could be ruled out for each of these events.
During the course of the study, two AIDS-defining events occurred in the ATV/LPV/r arm, one Pneumocystis pneumonia and a case of Non-Hodgkin Lymphoma 20 days and three months after baseline visit, respectively. An association between the AIDS-defining events and study drugs could not be established. One additional patient in the ATV/LPV/r arm was diagnosed with progressive multifocal leukencephalopathy (PML), but was retrospectively classified as having this condition prior to commencement of antiretroviral therapy, based on his neurological symptoms. None of the patients died during the study. No AIDS-defining event was observed in the CBV/LPV/r arm.
The primary endpoint of this study, metabolic side effects, could not be evaluated since the study was terminated prematurely. The quality of life data will be published elsewhere.
DISCUSSION
Our study was originally laid out to examine metabolic effects as well as quality of life of two different therapy regimens in HIV-infected, therapy-naive patients and to compare with each other. 80 of 100 originally planned patients were enrolled into the study and alternating assigned either to the double-PI arm including ATV/LPV/r (Reyataz® and Kaletra®) or to conventional CBV/LPV/r (Combivir® and Kaletra®) in the control group.
In an interim analysis, 24-week data were evaluated for virologic and immunological course of a total of 48 patients. In the study protocol the virologic failure was defined as a virus load above 50 copies HIV-RNA/mL by week 48.
Several factors may influence the virologic response to antiretroviral treatment, in particular potency, tolerability, and adherence [7,9]. Regarding tolerability, more patients developed gastrointestinal adverse events in the CBV/LPV/r arm, whereas hyperbilirubinemia was only seen in the ATV/LPV/r arm. Considering that pharmacokinetic measurements revealed no significantly reduced protease inhibitor levels in patients with failing dual PI regime this would not argue for reduced adherence as underlying cause. However, one should consider that possible adherence issue were not specifically captured, since we did not evaluate adherence by either patient self-report of adherence, pill counts or electronic measurement devices. It is important to note that the intend-to-treat analysis virological failure as end point considers all patients that stopped medication for whatever reason. Although this restrictive estimation is preferred in many clinical trails, since it gives a better assessment of the clinical efficacy of the treatment regimens, it can be misleading in terms of the virological reasons for treatment failures with either increase in viral load or resistance mutation development. This is particularly important, given that our study medication was not blinded, which could lead to even further bias in adverse event reporting and viral efficacy judgement. This is also relevant when looking at the “as-treated” analysis. This examination ignores all patients discontinuing because of insufficient virological efficiency and therefore tends to overestimate the virologic potency. As can be seen in our results, an “as-treated” analysis considerably reduced the differences in response rates between both arms. We wish to point out that in the ATV/LPV/r arm 11 patients switched therapy because of true virological failure and conclude from this that the dual PI arm performed definitely inferior to the CBV/LPV/r arm in terms of achieving a viral load below 50 copies/mL.
Due to antiviral potency, the NRTI-sparing regimen of atazanavir and ritonavir boosted lopinavir demonstrated less virologic efficacy than the conventional NRTI-based regimen in this study. The mechanism is not clear, but most likely at least two classes of antiretroviral drugs are required particularly in the initial treatment of HIV-1 infection, when viral replication is high [10-13]. Potentially, co-administration of two protease inhibitors is not beneficial or synergistically active due to competitive effector mechanisms. Similar results were found in the 2IP ANRS 127 trial, where two different PI-combinations showed delayed virologic response in therapy-naïve patients [14]. Furthermore, the MONARK trial demonstrated lower rates of virologic suppression in lopinavir/ritonavir monotherapy compared with lopinavir/ritonavir and a conventional NRTI-backbone in therapy-naïve patients [15,16]. Although it has been shown that a NRTI-sparing regimen is more likely associated with drug resistance [7], an alternative explanation for the lower rate of complete virological response in the dual PI-arm could be that these drugs act later in the virus life cycle. Using a mathematical model to investigate the effects of various drug classes on the dynamics of HIV-1 decay, Sedaghat et al. [17] have shown that the stage at which a drug acts affects the dynamics of viral decay. They predicted that the drug class acting latest in the viral life cycle dictates the dynamics of HIV-1 decay and found that the later in the life cycle an inhibitor acts, the more rapid the decay in viremia was.
We wish to emphasize that several points should to be considered in regard to the resistance testing. Our study is limited by the fact that we had only small numbers of sample available for successful resistance mutation analysis. Previous trails have shown that patients may develop viral rebound or fail to achieve a viral load below 50 copies in regimens containing PIs without viral resistance mutations. Although this can be considered as an advantage, since there is frequently no resistance mutation detectable in regions encoding for the protease as well as other relevant regions encoding for reverse transcriptase, this does not exclude resistance mutation development in regions of the virus that are not routinely assessed. Second, in only three out of seven patients with detectable viral load in the double-PI arm we were able to amplify sufficient material for sequence analysis and can not make firm conclusions on the overall resistance mutation development. Third, the detection of vial RNA could reflect release of HIV-RNA from latently infected cells and does not necessarily correlate to viral evolution and increasing viral replication. Finally, it could be possible that the detectable viremia is a result of release of HIV-RNA from pharmacologically “sanctuary sites” such as the central nervous system, where complete suppression is not achieved for pharmacological reasons. We do not know, whether the HIV-1 replicative capacity is reduced in our patients with virologic failure and persistent LLV, as suggested by other studies [17-19]. The extent of the decline in viral fitness, however, may be limited as HIV may eventually evade the selective pressure of a drug [20].
Nevertheless, dual PI regimen could be evaluated as an option in patients for a maintenance regimen. Consistent with this option, LPV/r mono-therapy in the OK-04 study showed that lopinavir/ritonavir mono-therapy can maintain HIV viral suppression in a very large proportion of patients [21] and similar outcomes were obtained with darunavir/r as single agent [22]. However, this may not apply for all protease inhibitors and depend on pretreatment [23]. The different immunological response with greater increase in CD4 cells in our study may be explained by myelosuppressive mechanisms by zidovudine. Furthermore, antiapoptotic effects on CD4 cells have been suggested for HIV-1 protease inhibitors, which are independent of the antiviral effects [24].
The previous objectives of this study to examine the effect of a NRTI sparing regimen on toxicity and metabolic parameters versus a NRTI containing regimen could not be reached due to early discontinuation. Once it became evident that the double-PI regimen of ATV/LPV/r was inferior with regard to virologic response compared to CBV/LPV/r, we stopped recruitment of study patients. Patients with virological failure were switched to the CBV/LPV/r regimen or another standard ART regimen.
In summary, our study revealed that the dual PI regimen comprising ATV/LPV/r had less virologic efficacy in an intent-to-treat analysis of therapy naïve HIV-patients than the conventional RTI-based regimen. As already shown by Landman et al. [14] dual protease inhibitor regimens were insufficient to rapidly suppress plasma HIV-RNA to less than 50 copies/mL in antiretroviral-naïve patients. Therefore protease inhibitor mono-therapy strategies may be useful rather for simplification of HAART in patients with suppressed HIV-RNA.
DISCLOSURES
M.S. Received honoraria as a lecturer by Abbott, Bristol-Myers-Squibb, Boehringer, Ingelheim, Glaxo-Smith-Kline, Roche, Gilead, Tibotec, Pfizer, Merck, Sharp & Dohme, and DuPont. He had served as a scientific advisor for Glaxo-Smith-Kline, Gilead, DuPont, Merck, Sharp & Dohme, Abbott, Roche, Solvay, and Tibotec. He received funding for scientific projects by Boehringer-Ingelheim, Glaxo-Smith-Kline, Roche, and Tibotec
B.S. Received speakers honorarium from GSK, Gilead, BMS, consulting fees from GSK, Gilead, BMS, Abbott, and educational grants from all
G.B. Reports that he has served on advisory boards and/or as speakers for and/or received research grants from Abbott, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Science, GlaxoSmithKline, Merck, Pfizer, Theratechnologies, and Tibotec.
R.E.S. Reports that he has served on advisory boards and/or as speakers for and/or received research grants from Abbott, Boehringer Ingelheim, Bristol-Myers Squibb, Pfizer and Tibotec.
ACKNOWLEDGEMENT
This study was supported by Abbott Corporation.

