All published articles of this journal are available on ScienceDirect.

Efficacy and Safety of Abacavir/Lamivudine/Zidovudine Plus Tenofovir in HBV/HIV-1 Coinfected Adults: 48-Week Data
Authors Info & Affiliations
Abstract
In HBV/HIV-coinfected patients, the risk of end-stage liver disease and death is increased. This open-label, prospective, pilot study evaluated abacavir/lamivudine/zidovudine twice daily plus tenofovir once daily in HBV/HIV-coinfected antiretroviral-naïve subjects. Nine adults (8 males) enrolled, with baseline mean HIV-1 RNA = 4.5 log10 copies/mL, HBV DNA = 9.0 log10 copies/mL, and median CD4 count =158 cells/mm3. No subject had baseline ALT >5x ULN.
Six subjects completed the study: 1 withdrew due to non-treatment-related toxoplasmosis and 2 were lost-to-follow-up. At week 48, 100% (6/6) of remaining subjects had ≥2 log10 decrease in HBV DNA, and 100% (6/6) and 83% (5/6) had HIV-1 RNA <400 and <50 copies/mL, respectively. Median change from baseline in CD4 count was 157 cells/mm3. One subject experienced treatment-related grade 3 leukopenia. These results demonstrate that abacavir/lamivudine/zidovudine and tenofovir were well tolerated with sustained HIV-1 and HBV antiviral activity through 48 weeks in HBV/HIV-coinfected, antiretroviral-naïve subjects.
DEAR EDITOR:
AIDS-related morbidity and mortality have declined as a result of highly active antiretroviral therapy (HAART); however, liver disease caused by chronic hepatitis B virus (HBV) infection remains an important cause of morbidity and mortality among HIV-infected patients [1,2]. In HBV/HIV-coinfected patients, the risk of end-stage liver disease and death is increased, and liver histological damage may progress more rapidly leading to cirrhosis in a shortened timeframe [3].
Current treatment guidelines for HBV/HIV coinfected patients recommend lamivudine (3TC) or emtricitabine in combination with tenofovir (TDF) to potentially reduce the risk of HBV resistance associated with 3TC monotherapy [4]. Abacavir/3TC/zidovudine (ABC/3TC/ZDV; Trizivir, GlaxoSmithKline, Research Triangle Park, North Carolina) is a fixed-dose combination product containing three nucleoside reverse transcriptase inhibitors (NRTIs): 300 mg ABC, 150 mg 3TC, and 300 mg ZDV, for the treatment of HIV infection. ABC/3TC/ZDV is administered as 1 tablet taken twice daily and has been shown to facilitate treatment adherence [5,6]. The clinical safety and efficacy of ABC/3TC/ZDV has been demonstrated in several clinical trials. TDF is a nucleotide reverse transcriptase inhibitor approved for the treatment of HIV infection in combination with other agents at a recommended dose of 300 mg (1 tablet) once daily. TDF is approved for hepatitis B therapy, and data from clinical trials have confirmed the activity of TDF in both 3TC-naïve and 3TC-resistant HBV patients [7,8].
In this open-label, prospective, pilot study, we evaluated the long-term efficacy and safety of ABC/3TC/ZDV twice daily coadministered with TDF once daily in HBV/HIV-coinfected antiretroviral-naïve subjects. At the time this study was designed, ABC/3TC/ZDV was recommended by the DHHS Panel on Clinical Practices for Treatment of HIV Infection as an alternative regimen in treatment-naive patients [9], and data on the use of TDF in this population had only recently become available [10]. In that context, this regimen provided a straightforward dosing regimen uncomplicated by food or fluid restrictions and a low daily pill burden, and it is also nucleoside reverse transcriptase inhibitors (NNRTI)- and protease inhibitor (PI)- sparing to preserve future HIV treatment options.
Nine subjects with documented HIV-1 and HBV infection were enrolled at 5 centers in the United States between October 2003 and March 2005. Inclusion criteria included subjects who were ART-naïve, older than 18 years of age, had a CD4 count ≥100 cells/mm3, a plasma HIV-1 RNA count ≥1000 copies/mL, a HBV DNA viral load >1,000,000 copies/mL, and were HBsAg positive, HBsAb negative, HBeAg positive, and HBeAb negative. Subjects were excluded if they had an AIDS-defining illness within 30 days of screening. All subjects provided written informed consent to participate, and the protocol for the study was approved by the institutional review boards for each study site.
At each study visit, patients were monitored for adverse events and laboratory abnormalities using routine hematology and clinical chemistry panels that were processed at a central laboratory facility. Medication adherence was documented using pill counts, and measurements of HBV DNA, HIV-1 RNA, and CD4 cell counts were made throughout the study. HIV-1 RNA levels were determined using the Roche Amplicor HIV-1 Monitor COBAS UltraSensitive test (version 1.5; range of detection, 51-75,000 copies/mL) and the standard test (range of detection, 400-750,000 copies/mL). HBV DNA levels were measured using an independently validated real-time PCR assay performed at a central laboratory. Genotypic analyses of baseline HIV and HBV isolates were determined using the TRUGENE HIV-1 Genotyping Kit (Bayer HealthCare LLC, Tarrytown, NY) and an independently validated polymerase chain reaction/sequencing assay (Quest Diagnostics Nichols Institute, San Juan Capistrano, CA), respectively. Phenotypic analysis of baseline HIV isolates was determined using the PhenoSense GT assay (Monogram Biosciences, South San Francisco, CA).
HIV-1 virologic failure was defined as HIV-1 RNA ≥400 copies/mL at week 24 or later that was confirmed by a second measurement of HIV-1 RNA ≥400 copies/mL less than 4 weeks after the first measurement. Confirmed HBV virologic failure was defined as a <2 log10 copies/mL decline in HBV at week 24. The primary efficacy endpoint was proportion of subjects with a ≥2 log10 copies/mL decrease in HBV DNA viral load at week 48. The analysis population included all enrolled subjects that consumed at least one dose of study drug.
Of the 9 subjects enrolled, 8 were male and 1 was female. Three subjects were white, 2 were black, and 4 were American Hispanic. The mean age ±standard deviation (SD) was 39.4 ±11.06 years. At baseline, the mean ±SD HBV DNA viral load was 9.0 ±0.6 log10 copies/mL and the mean ±SD HIV-1 RNA viral load was 4.5 ±0.77 log10 copies/mL. The median (range) CD4 cell count was 158 cells/mm3 (97–596 cells/mm3). The median (range) alanine aminotrans-ferase (ALT) level was 55 IU/L (30–144 IU/L). No subject had abnormal (>5 times the upper limit [48 IU/L] of the normal range) ALT levels at baseline. No HIV-1 NRTI mutations were detected in any of the 9 subjects at baseline. Likewise, no mutations were observed at YMDD/M552 or FLLAQ/L528 in the HBV polymerase from any of the 9 subjects at baseline.
Six subjects completed the study and 3 subjects discontinued the study before week 48: 1 withdrew because of a medical condition unrelated to study drug, and 2 were lost to follow-up. None of the subjects experienced protocol-defined HBV virologic failure. One subject (HIV-1 RNA 740 copies/mL at week 24) was an unconfirmed HIV-1 virologic failure (patient did not return after week 24 visit); no other subject experienced protocol-defined HIV-1 virologic failure.
The time course of the change from baseline in HIV-1 RNA levels for all 9 subjects is shown in Fig. (1A). At week 48, 100% (6/6) and 83% (5/6) of subjects had plasma HIV-1 viral load <400 copies/mL and <50 copies/mL, respectively. The mean change from baseline in HIV-1 RNA levels at week 48 was -2.9 log10 copies/mL. The median (range) CD4 cell count at week 48 was 356 cells/mm3 (179–695 cells/mm3) and the median (range) change from baseline in CD4 cell counts was 157 cells/mm3 (62–253 cells/mm3).
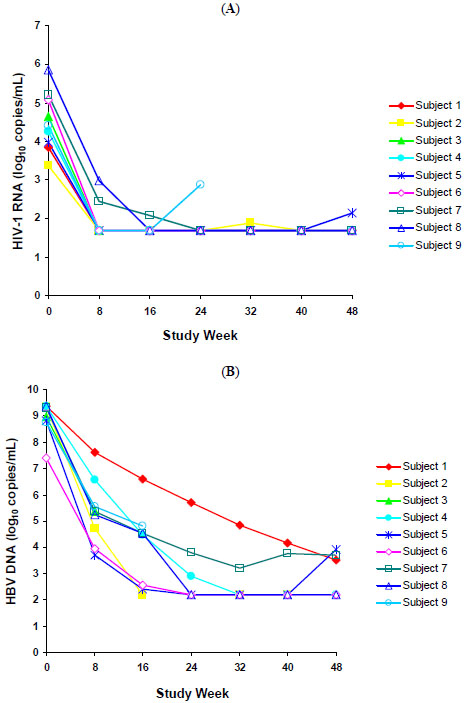
(A) HIV-1 RNA levels for nine HBV/HIV-coinfected antiretroviral-naïve subjects treated with ABC/3TC/ZDV + TDF over 48 weeks. (B) HBV DNA levels for nine HBV/HIV-coinfected antiretroviral-naïve subjects treated with ABC/3TC/ZDV + TDF over 48 weeks.
Fig. (1B) shows the time course of the change from baseline in HBV DNA levels for all 9 subjects. At week 48, 100% (6/6) of subjects had a ≥2 log10 copies/mL decrease in HBV DNA viral load, and the mean ±SD decrease in HBV DNA viral load was -6.0 ±0.98 log10 copies/mL. One subject achieved HBeAg seroconversion at week 48. This subject’s HBV DNA level and HIV-1 RNA viral load decreased from 8.8 and 4.0 log10 copies/mL at baseline to 3.9 and 2.1 log10 copies/mL, respectively, at week 48. The subject’s ALT level normalized to 11 IU/L from a baseline value of 55 IU/L, and the CD4 cell count increased from 114 cells/mm3 at baseline to 179 cells/mm3 at week 48.
Three subjects each experienced a grade 3 adverse event that was unrelated to study drug: decreased neutrophil counts (1 subject), increased lipase (1 subject), and toxoplasmosis (1 subject who withdrew from the study). A fourth subject, who experienced drug-related grade 3 leukopenia that remained unresolved, continued on study. Three subjects had abnormal ALT levels during the study.
The results from this prospective pilot study demonstrated that treatment with ABC/3TC/ZDV + TDF was well tolerated with sustained antiviral activity through 48 weeks against HBV and HIV-1 in HBV/HIV-coinfected antiretroviral-naïve subjects. The virological efficacy of ABC/3TC/ZDV + TDF in HBV/HIV coinfected antiretroviral-naïve subjects is consistent with the results reported by Dore, et al. [8]. During 48 weeks of therapy with ABC/3TC/ZDV + TDF, a mean ±SD decrease of 6.0 ±0.98 log10 copies/mL in HBV DNA was observed in this study whereas a mean decrease in HBV DNA of 4.7 log10 copies/mL was observed by Dore, et al. in antiretroviralnaïve subjects who received antiretroviral therapy regimens containing 3TC and TDF. In this study, HBeAg seroconversion occurred in 1 subject who had good suppression of HBV DNA with normalization of ALT, but little increase in CD4 cells over 48 weeks.
The ABC/3TC/ZDV + TDF regimen is a twice-daily regimen that requires the subject to take 1 tablet of ABC/3TC/ZDV twice daily and 1 tablet of TDF once daily. This pilot study indicated that this regimen provided durable suppression of HIV-1 and HBV in HBV/HIV coinfected antiretroviral-naïve subjects. Importantly, as a NNRTI- and PI-sparing regimen, the possibility of preserving future options for HIV-1 treatment with this regimen remains. Current treatment guidelines either do not recommend this single-class regimen of ABC/3TC/ZDV+ TDF for initiation of HIV therapy due to limited data [11] but indicate that this regimen can be “considered in special circumstances” [12] as it may represent a viable option for those patients in whom treatment with other conventional regimens may not be feasible due to drug interaction or tolerability issues. Examples of these special circumstances include women of childbearing potential and patients with baseline psychiatric illnesses requiring treatment with other medications that have substantial drug interactions with NNRTIs and/or PIs.
AUTHOR DISCLOSURES
Dr. Rodriguez receives honoraria from Abbott Laboratories, Gilead Sciences, Pfizer, and Tibotec. Dr. DeJesus receives research support or honoraria from Abbott Laboratories, Boehringer Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Hoffman LaRoche Laboratories, Merck, Pfizer, Schering Plough, Tibotec, and Vertex Pharmaceuticals. Vanessa Williams, David Irlbeck, Lisa Ross, and Belinda Ha, PhD, are currently employees of GlaxoSmithKline. Charles Lancaster was an employee of GlaxoSmithKline at the time the study was conducted.
ACKNOWLEDGEMENTS
This investigator-initiated research was funded by GlaxoSmithKline. We thank Terry Paul, PhD, and Katherine A. DeBruin, PhD, for their writing and editorial assistance in the preparation of the manuscript. We also thank Britt Stancil for statistical support and Bert Torres for programming support.

